Coordenada intrínseca de reação – reação E2
dezembro 3, 2015 Deixe um comentário
Olá
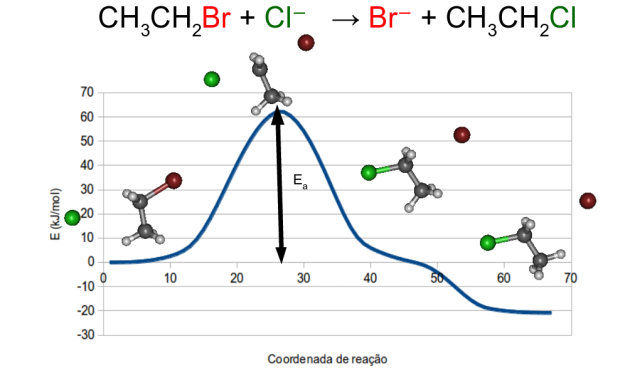
Segue o vídeo da coordenada intrínseca de reação (IRC) para uma eliminação bimolecular (E2) entre o 2-cloro-propano e o íon hidroxila:
CH3CHClCH3 + OH– → CH3CH=CH2 + H2O + Cl–
Ela foi calculada com o Firefly 8.0.1 com a base 6-31G(df) incluindo funções difusas seguindo o procedimento detalhado no tutorial para as reações SN2: https://kalilbn.wordpress.com/tutorial-firefly-gabedit/tutorial-calculo-de-estado-de-transicao-e-irc/
Essa mesma molécula orgânica poderia também sofrer um ataque do tipo SN2 por parte do íon OH⁻ caso esse reagisse com o carbono ao qual está ligado o cloro. Na reação E2, entretanto, o íon OH⁻ atua como base extraindo um próton e simultaneamente temos a saída do íon cloreto formando uma ligação dupla C=C no produto. Após a reação, há uma reorganização a fim de favorecer a interação entre a molécula de água e o íon cloreto (em fase condensada isso não ocorreria pois o cloreto já seria solvatado pelas moléculas de solvente próximas ao sair).
Assim como discutido no caso da reação SN2, também na reação E2 o orbital HOMO no estado de transição apresenta caráter antiligante com relação as ligações formadas ou rompidas, no caso, entre o hidrogênio do substrato e o oxigênio do íon OH⁻ e entre o carbono e o cloro, além disso, já nota-se alguma semelhança na região entre os dois átomos de carbono com o orbital π que é o orbital HOMO do alceno resultante da reação.
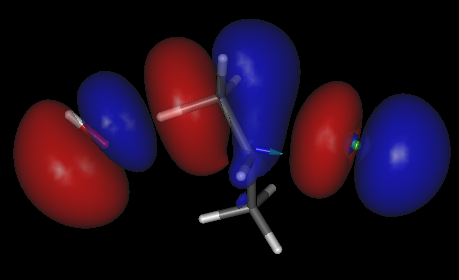
HOMO do estado de transição para uma reação E2
Os arquivos de input e output para otimização do estado de transição dessa reação estão disponíveis pelo diretório do blog no Google Drive: https://drive.google.com/open?id=0BxbfUlGvt1wJQmN2eVZBSmNMVDA
Referências dos softwares e funções de bases usados: