Tutorial LAMMPS – mistura de água e etanol – parte 1
Tutorial LAMMPS – mistura de água e etanol
Página 1 – preparando e rodando a simulação
- Introdução
- Softwares necessários
- Preparação dos arquivos de input
- Descrição dos arquivos de input
- Execução da simulação
- Visualização da trajetória
Página 2 – análises dos resultados
- Componente de energia e densidade
- Convertendo a trajetória para pdb
- Distribuição radiais de pares (rdf)
- Distribuição espacial (sdf)
- Distribuição de diedros
- Problemas propostos
- Referências
.
1. Introdução
Nesse primeiro tutorial de dinâmica molecular com o programa LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) serão apresentadas ferramentas gratuitas para produzir seus arquivos de input e para análise de resultados. Apesar de ser a mesma metodologia, a estrutura de input do LAMMPS é bastante diferente da estrutura de input do Gromacs, abordado em outros tutoriais nesse blog. Como exemplo, trabalharemos nesse tutorial com uma mistura equimolar de água e etanol. Diversos outros exemplos de aplicações com o LAMMPS, especialmente envolvendo sólidos, podem ser encontrados na página https://icme.hpc.msstate.edu/mediawiki/index.php/LAMMPS_tutorials .
O etanol, também conhecido como álcool etílico, C2H5OH, é um composto orgânico miscível com a água em qualquer proporção. Tanto o álcool vendido comercialmente em supermercados quanto em postos de combustível para abastecimento de veículos são na verdade misturas de etanol e água, sendo a fração de água no álcool combustível menor do que no álcool vendido para aplicações domésticas. É comum o uso da unidade ºGL (grau Gay Lussac) para indicar o teor de álcool em misturas com água, essa unidade indica a fração em volume de álcool. Sendo as massas molares da água e do etanol 18,02 e 46,08 g/mol e suas densidades 1,0 e 0,79 g/cm³, respectivamente, obtém-se os volumes molares dos líquidos puros como 18,0 e 58,3 ml/mol, de modo que uma mistura equimolar como a que trabalharemos nesse tutorial corresponda a 76ºGL, o que é aproximadamente a composição usada para esterilização em aplicações domésticas.
Nesse tutorial iremos preparar uma caixa de simulação contendo 300 moléculas de etanol e 300 moléculas de água usando para isso os programas fftool e packmol. Realizaremos em sequência uma simulação no ensemble NPT (isso é, número de partículas, pressão e temperatura constantes), extrairemos o volume médio dessa e faremos então uma simulação a volume constante (NVT). Na segunda parte desse tutorial trabalharemos com a análise dos resultados, usando o programa TRAVIS para realização de algumas análises estruturais. A sessão 4 apresenta uma discussão mais detalhada sobre os arquivos de input e pode ser pulada por quem estiver interessado apenas em realizar a simulação e fazer as análises.
Veja também as paginas sobre conceitos fundamentais de dinâmica molecular e outros tutoriais de dinâmica molecular:
- Dinâmica molecular – conceitos fundamentais
- Tutorial GROMACS – água
- Tutorial GROMACS – íons em água
- Tutorial GROMACS – moléculas em solução
.
.
2. Softwares necessários
As simulações de dinâmica molecular serão executadas com o pacote LAMMPS, o mesmo pode ser baixado diretamente do repositório de softwares em diversas distribuições Linux, procure por ele no gerenciador de softwares da sua distribuição ou instale via terminal, no caso de distribuições com suporte a pacotes debian, por meio dos comandos:
sudo add-apt-repository ppa:gladky-anton/lammps
sudo apt-get update
sudo apt-get install lammps-daily
Também é possível baixar binários ou o código fonte diretamente do site do LAMMPS tanto para outras distribuições Linux quanto para Windows ou Mac. Verifique as diversas opções no site https://lammps.sandia.gov/doc/Install.html
Para a preparação da estrutura inicial e dos arquivos de input, será utilizado o programa fftool, disponível em https://github.com/agiliopadua/fftool, e o programa Packmol, disponível em http://m3g.iqm.unicamp.br/packmol/home.shtml. O Packmol, desenvolvido por pesquisadores da USP e da Unicamp, é um programa bastante versátil que permite, a partir das estruturas das moléculas e íons, preparar caixas de simulação não apenas com misturas homogêneas, como faremos aqui, mas também permite produzir estruturas iniciais mais complexas como membranas e vesículas. Além disso, as estruturas são geradas em formatos comuns, como xyz e pdb, e podem ser usadas como arquivos de input (ou convertidas em arquivos de inputs) para outros programas de dinâmica molecular, Monte Carlo, ou mesmo de química quântica. O Gromacs, por exemplo, permite trabalhar diretamente com estruturas em formato pdb. O fftool, desenvolvido por Agílio Pádua, da Universidade de Lyon, gera os arquivos de input propriamente ditos convertendo a estrutura gerada pelo Packmol no formato lido pelo LAMMPS, no qual, conforme veremos, parâmetros ligantes e cargas parciais são descritas no mesmo arquivo que contém as coordenadas iniciais dos átomos, o que é uma diferença significativa em relação ao Gromacs onde tais informações são especificadas em arquivos de input separados.
A compilação do Packmol em ambiente Linux é bastante simples. Após baixar o arquivo .tar.gz, execute os seguintes comandos no diretório no qual ele se encontra para descompactá-lo e criar os executáveis:
tar -xvzf packmol.tar.gz
cd packmol
make
Caso encontre problemas, siga as instruções fornecidas no manual do programa (http://m3g.iqm.unicamp.br/packmol/userguide.shtml). Caso tenha acesso de administrador e achar conveniente, copie o executável packmol gerado para o diretório /usr/bin, assim, poderá usá-lo diretamente em qualquer diretório usando apenas o comando “packmol”. Caso contrário, será necessário copiá-lo para o diretório ou especificar o caminho completo toda vez que for usá-lo. O programa fftool não necessita de compilação.
Um programa em python escrito por mim será fornecido para extrair informações impressas no arquivo de log gerado pelo LAMMPS, tais como volume e energia do sistema. Para análises estruturais, será utilizado o programa TRAVIS, disponível em http://www.travis-analyzer.de/. Esse programa é interativo, apresentando opções em menus em modo texto e fazendo perguntas ao usuário para cada opção desejada, o que torna seu uso bastante simples e intuitivo. Ele possui diversas funções de análise, aqui usaremos para calcular distribuições radiais de pares, distribuições espaciais e distribuições de diedros. Cabe mencionar que distribuições radiais de pares e de diedros podem ser calculadas diretamente pelo LAMMPS no decorrer da simulação já especificando elas no arquivo de input, mas eu pelo menos considero o pós-processamento da trajetória em geral mais interessante que análises estruturais feitas ao longo da simulação pois frequentemente não saberá a priori quais análises estruturais serão relevantes para explicar tendências observadas no decorrer de um estudo com simulações.
A compilação do TRAVIS é similar a do Packmol: Extraia o conteúdo do arquivo compactado, entre no diretório do programa e execute “make”. Se desejar, pode alterar o compilador C++ alterando o arquivo Makefile. Será gerado o diretório “exe” dentro do qual estará o executável do TRAVIS. Assim como no caso do Packmol, pode ser conveniente copiá-lo para o diretório /usr/bin para poder executá-lo de qualquer diretório sem especificar o caminho completo.
Para visualização da trajetória e dos resultados de distribuição espacial, utilizaremos o programa VMD (Visual Molecular Dynamics), disponível em https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD. Esse mesmo programa é discutido nos tutoriais desse blog empregando o Gromacs. Para visualização das distribuições radiais de pares e componentes de energia, empregaremos o programa Grace, disponível no repositório das principais distribuições Linux, mas qualquer outro software que permita plotar gráficos a partir de um arquivo com duas colunas de dados pode ser usado, tal como o Gnuplot.
.
.
3. Preparação da simulação
Nesse tutorial usaremos arquivos de estruturas já prontos no diretório “examples” do fftool. De fato, o que faremos aqui é bastante similar ao exemplo do arquivo README do fftool, mas trabalharemos com uma composição diferente e faremos algumas alterações nos arquivos gerados.
Vá ao diretório “examples” do fftool. Para gerar os arquivos de input do LAMMPS são necessários arquivos com as estruturas de cada molécula em formato de matriz Z como a mostrada abaixo para o etanol (ethanol.zmat):
ethanol 1 C3H 2 CTO 1 1.47 3 H 2 1.09 1 109.5 4 H 1 1.09 2 109.5 3 180.0 5 H 2 1.09 1 109.5 4 60.0 6 H 1 1.09 2 109.5 5 180.0 7 OH 2 1.41 1 109.5 6 60.0 8 H 1 1.09 2 109.5 7 180.0 9 HO 7 0.945 2 109.5 1 180.0 oplsaa.ff
A primeira linha é apenas o nome da molécula. Da terceira linha até a antepenúltima temos as especificações dos átomos sendo a primeira coluna o número do átomo, a segunda o tipo atômico (usando o mesmo nome especificado no arquivo do campo de força). A terceira coluna é o número do átomo com o qual especificamos um comprimento de ligação, a quarta o valor do comprimento em Å, seguido pelo número de outro átomo para especificar um ângulo, o valor do ângulo, um quarto átomo para a especificação de um diedro e finalmente o valor do ângulo de diedro. Na última linha do arquivo é dado o nome do campo de força onde encontram-se os parâmetros de interação ligantes e não-ligantes de cada tipo atômico da molécula. No caso, usaremos os parâmetros do campo de força OPLS-AA (optimized parameters for liquid simulations – all atoms). Tomando como exemplo o átomo 7, que é oxigênio do etanol, ele tem tipo atômico “OH” e é especificada na matriz Z a distância dele ao átomo 2, o carbono “CTO” como 1.41 Å, o ângulo OH – CTO – CH3 como 109.5º e o diedro formado por esses átomos e um dos hidrogênios ligados ao carbono “CH3” como 60.0º. No arquivo do campo de força oplsaa.ff temos inicialmente as especificações dos parâmetros não-ligantes de cada tipo atômico, incluindo a carga parcial e os parâmetros σ e ε do potencial de Lennard-Jones, (cuja forma funcional é dada pela equação abaixo), seguidos pelos potenciais de ligações, ângulos e diedros.
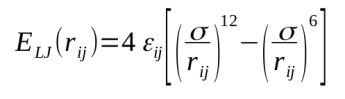
Queremos preparar uma caixa de simulação contendo 300 moléculas de etanol e 300 moléculas de água. Para isso, dentro do diretório examples, use o comando:
../fftool 300 ethanol.zmat 300 spc.zmat -b 36.0
Onde ethanol.zmat e spc.zmat são os arquivos com as estruturas em formato de matriz Z para o etanol e a água, respectivamente, já fornecidos junto com o fftool. A variável -b especifica a aresta inicial da caixa de simulação em Å. Como veremos, isso resulta em uma densidade menor do que a esperada para a mistura, mas trabalhar com uma densidade inicial um pouco mais baixa facilita o empacotamento aleatório das moléculas pelo Packmol e a densidade normalmente converge rapidamente para o valor de equilíbrio, pelo menos para líquidos puros e misturas simples como essa. Esse comando vai gerar o arquivo de input pack.inp para o Packmol. Nessa etapa, caso queira alguma estrutura diferente de uma mistura aleatória, por exemplo, desenhar um sistema bifásico ou começar com a moléculas em alguma orientação especifica, poderia fazê-lo mediante edições no arquivo pack.inp. Veja os exemplos na página do Packmol caso tenha interesse em gerar estruturas iniciais mais sofisticadas, para esse tutorial não será necessário editá-lo. Execute agora o Packmol:
packmol < pack.inp
Será gerado o arquivo simbox.xyz. Essa é a estrutura inicial do sistema e pode ser aberta no VMD ou em outro programa de visualização de estruturas moleculares (com o VMD, basta executar o comando vmd simbox.xyz). Uma representação dessa estrutura é dada abaixo.
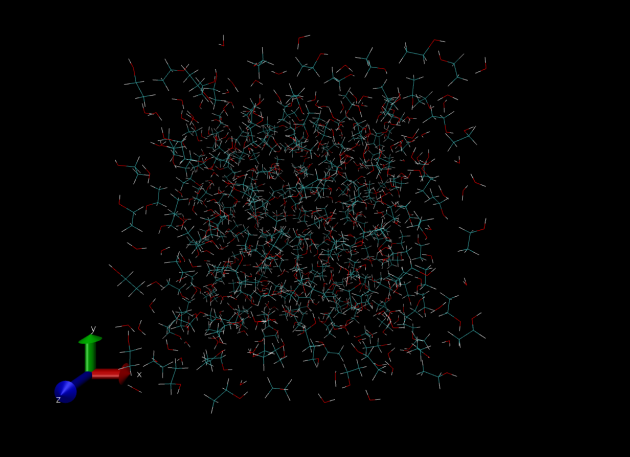
Figura 1 – Estrutura inicial para a simulação de água e etanol visualizada com o VMD.
O simbox.xyz, entretanto, não serve como arquivo de input para o LAMMPS pois ele contém apenas as coordenadas xyz e nomes atribuídos aos átomos enquanto o arquivo de entrada para o LAMMPS precisa também das massas e cargas parciais atômicas e especificação das ligações, ângulos e diedros e os respectivos parâmetros ligantes. Para gerar esse arquivo, executamos novamente o fftool agora acrescentando a opção -l (letra L minúscula) ao comando:
../fftool 300 ethanol.zmat 300 spc.zmat -b 36.0 -l
Ele irá agora ler o arquivo simbox.xyz gerado pelo Packmol para obter as coordenadas atômicas e os arquivos de campo de força (no caso, oplsaa.ff e spc.ff) para extrair os parâmetros de interação ligantes e não-ligantes. Serão gerados os arquivos data.lmp e in.lmp, vamos discutir brevemente a estrutura de cada um deles na próxima sessão. O leitor que desejar pular essa descrição pode baixar o arquivo in.lmp com as modificações descritas na sessão seguinte por meio do link abaixo e pular para a sessão 5.
.
.
4. Descrição dos arquivos de input
No arquivo data.lmp são especificadas as dimensões da caixa, cargas parciais, as massas atômicas, coordenadas cartesianas e parâmetros ligantes. Note aqui uma diferença para os arquivos de input do Gromacs: No LAMMPS, as cargas e parâmetros ligantes são incluídos no mesmo arquivo das coordenadas iniciais, enquanto no Gromacs são arquivos distintos. As primeiras linhas do data.lmp correspondem aos números totais de átomos, ligações, ângulos e diedros no sistema seguido pela quantidade de tipos diferentes de átomos, ligações, ângulos e diedros e pelas arestas da caixa de simulação. Na sequência observamos as massas molares de cada tipo atômico e os parâmetros para interações ligantes, cujas formas funcionais são dadas para o campo de força OPLS-AA nas equações abaixo.
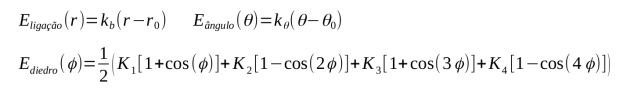
As ligações e ângulos são descritos por potenciais harmônicos, sendo que no arquivo data.lmp a primeira coluna atribui um número aquele tipo de ligação ou ângulo, a segunda coluna corresponde as constantes de força (kb ou kθ) e terceira ao valor de equilíbrio da ligação ou ângulo (r0 ou θ0 nas expressões dos potenciais). Para os diedros são especificados os 4 coeficientes Ki de acordo com a equação anterior.
O LAMMPS pode trabalhar com diferentes sistemas de unidades, sendo esse especificado no arquivo in.lmp e, no caso, usaremos o sistema “real”, onde a energia é dada em kcal/mol, a massa em g/mol, temperatura em K, pressão em bar e distâncias e coordenadas cartesianas em Å. Desse modo, a constante de força das ligações tem seu valor dado em kcal mol-1 Å–2, por exemplo. Para mais detalhes sobre os sistemas de unidades do LAMMPS, consulte a página https://lammps.sandia.gov/doc/units.html.
Após os coeficientes das interações ligantes, temos as especificações de cada átomo no sistema na diretriz “Atoms”. A primeira coluna é o número do átomo, a segunda o número da molécula a qual o átomo pertence, a terceira é o tipo do átomo (deve corresponder aos tipos na diretriz “Masses”), a quarta é a carga parcial e as 3 colunas seguintes as coordenadas cartesianas na estrutura inicial. Note que fftool acrescenta ainda o nome atribuído no campo de força e na matriz Z a cada átomo como comentário no final de cada linha.
Após a diretriz Atoms, temos as listas de todas as ligações, ângulos e diedros definidos no sistema. O primeiro número nessas listas é apenas um contador, o segundo atribui os coeficientes definidos anteriormente para a interação e os números seguintes são os números dos átomos envolvidos na interação (2 para ligações, 3 para ângulos e 4 para diedros).
Discutiremos a seguir o arquivo in.lmp, onde são especificadas condições de simulação, parâmetros de van der Waals e, opcionalmente, análises a serem feitas ao longo da simulação. Discutiremos passo a passo ele destacando em vermelho linhas que acrescentei ou fiz alguma modificação em relação ao gerado pelo fftool. O arquivo completo pode ser baixado no link no final dessa sessão. Não pretendo discutir todas as opções envolvidas em cada comando, para uma descrição detalhada sobre cada um, busque pelo comando respectivo na página do LAMMPS. As primeiras linhas são:
# created by fftool
units real
boundary p p p
A primeira linha do arquivo in.lmp após o comentário inserido pelo fftool especificam o sistema de unidades usado, no caso “real”. A segunda linha define o uso de condições periódicas de contorno nas 3 direções cartesianas. Devido às condições periódicas de contorno, uma molécula próxima a uma beirada da caixa interage com as moléculas do lado oposto da caixa e, do mesmo modo, se a molécula atravessar uma das faces da caixa, isso é equivalente a ela entrar na caixa pelo lado oposto. Como resultado, por mais que nosso sistema tenha um número bastante limitado de moléculas, ele se comporta de modo similar a um líquido bulk, não havendo nenhuma interface real com o vácuo, como mostra a representação a seguir (Figura 2) de uma estrutura extraída da simulação, onde a caixa real de simulação é mostrada colorida e suas réplicas periódicas em 2 direções são representadas em cinza. Para cada molécula do sistema, é como se estivesse envolta o tempo todo por outras moléculas de água e etanol independente de estar no centro ou nas bordas da caixa de simulação. Um exercício proposto no final desse tutorial servirá para mostrar o que ocorre se não empregarmos condições periódicas de contorno.

Figura 2 – Ilustração das condições periódicas de contorno, onde as moléculas reais estão representadas coloridas enquanto as réplicas periódicas nas direções x e y são representadas em cinza. Uma das moléculas de etanol foi destacada com representação de van der Waals. Note que as réplicas dessa molécula, também em destaque, apresentam exatamente a mesma estrutura e orientação e tem a posição deslocada por múltiplos de comprimentos das respectivas arestas da caixa de simulação.
As linhas seguintes indicam que os átomos possuem tanto interações eletrostáticas quanto de van der Waals e os tipos de potenciais usados para ligações, ângulos e diedros (os mesmos que discutimos anteriormente). A linha “special_bonds” indica que as interações não ligantes não são calculadas para átomos que estão a uma ou duas ligações de distância e que são escalados por 0,5 para átomos a 3 ligações de distância. Para átomos mais distantes do que isso dentro da mesma moléculas as interações não-ligantes são calculadas normalmente.
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style opls
special_bonds lj/coul 0.0 0.0 0.5
O próximo conjunto de linhas está relacionado às interações não ligantes, especificando em pair_style os raios de corte como 12.0 Å para ambas as interações não-ligantes. Note que as várias linhas pair_coeff especificam os parâmetros σ e ε do potencial de Lennard-Jones apenas para interações entre átomos do mesmo tipo. As interações entre tipos distintos são definidas com base em regras de combinação, sendo essas especificadas pela linha pair_modify, sendo no caso tomada a média geométrica entre os parâmetros das interações A-A e B-B para calcular os parâmetros das interações A-B. “Tail yes” implica no uso de correção de longo alcance para as interações de van der Waals (essa correção não afeta as forças no sistema, é apenas um fator aditivo na energia potencial total). A linha kspace_stype define as correções de longo alcance para as interações eletrostáticas, sendo usado para tal o algoritmo PPPM (Particle-Particle Particle-Mesh). O comando read_data indica o nome do arquivo com a estrutura inicial, no caso, data.lmp. Note a linha abaixo comentada com ‘#’ (esse símbolo significa que a linha não será processada pelo LAMMPS), onde temos o comando “read_restart”. O mesmo é usado para reiniciar ou estender uma simulação no LAMMPS através de um arquivo de restart gerado. Se for fazer isso, entretanto, execute a nova simulação em um diretório novo. Diferente do Gromacs, por exemplo, o LAMMPS sobrescreverá os arquivos antigos mesmo que a simulação nova tenha sido iniciada por meio de um restart se iniciá-la no mesmo diretório.
pair_style hybrid lj/cut/coul/long 12.0 12.0
pair_modify mix geometric tail yes
kspace_style pppm 1.0e-5
read_data data.lmp
# read_restart restart1.lmp
pair_coeff 1 1 lj/cut/coul/long 0.065999 3.500000 # C3H C3H
pair_coeff 2 2 lj/cut/coul/long 0.065999 3.500000 # CTO CTO
pair_coeff 3 3 lj/cut/coul/long 0.030000 2.500000 # H H
pair_coeff 4 4 lj/cut/coul/long 0.170000 3.120000 # OH OH
pair_coeff 5 5 lj/cut/coul/long 0.000000 0.000000 # HO HO
pair_coeff 6 6 lj/cut/coul/long 0.000000 0.000000 # Hw Hw
pair_coeff 7 7 lj/cut/coul/long 0.155425 3.165500 # Ow Ow
Logo após temos mais linhas escritas como comentários pelo fftool com os comandos para fazer minimização de energia e para reiniciar a contagem de passos. Como veremos, é possível fazer múltiplas simulações em sequência com um único arquivo de input do LAMMPS. Se quiser que cada uma inicie a contagem de passos em zero ao invés de continuar a contagem do fim da anterior, basta usar entre elas o comando reset_timestep 0.
# minimize 1.0e-4 1.0e-6 100 1000
# reset_timestep 0
A próxima linha indica o uso do algoritmo shake para restringir as ligações envolvendo átomos de hidrogênio, que apresentam elevadas frequências vibracionais, além do ângulo H-O-H na molécula de água. A linha neighbor especifica qual distância além do raio de corte deve ser usada para montar as listas de vizinhos. Como o raio de corte é 12,0 Å, a lista de vizinhos inclui partículas até 12,0 + 2,0 = 14,0 Å de distância. A linha neigh_modify comentada pode ser útil para simulações com instabilidades numéricas que resultem em erros do tipo “Missing atom … in bond …” ou “Missing atom … in angle …”. Logo em seguida é especificado o passo de integração dt como 1,0 fs.
fix SHAKE all shake 0.0001 20 0 b 2 4 5 a 6
neighbor 2.0 bin
# neigh_modify delay 0 every 1 check yes
timestep 1.0
As linhas seguintes são exemplos de definições de variáveis no LAMMPS, seguindo a estrutura “variable” + (nome da variável) + “equal” + (valor da variável ou expressão para calculá-la). Tais variáveis podem receber um valor numérico constante ou podem ser expressões matemáticas envolvendo outras variáveis, como veremos a seguir (veja a página https://lammps.sandia.gov/doc/variable.html para mais detalhes). Essas duas variáveis correspondem aos valores de temperatura e pressão em que realizaremos a simulação, 300 K e 1 bar, respectivamente. A vantagem de especificá-los como variáveis é que, se formos usar o mesmo valor em diversos comandos no arquivo de input, só precisamos modificá-lo uma única vez se pretendermos, por exemplo, fazer uma simulação em outra temperatura. O comando velocity cria as velocidades iniciais com a distribuição esperada para a temperatura de 300 K.
O comando seguinte, fix npt, define o ensemble da simulação como sendo NPT (número de partículas, pressão e temperatura constantes). Esse comando emprega o termostato e o barostato de Nosé-Hoover. Para mais detalhes sobre termostatos e barostatos disponíveis no LAMMPS, veja as páginas https://lammps.sandia.gov/doc/Howto_thermostat.html e https://lammps.sandia.gov/doc/Howto_barostat.html .
variable TK equal 300.0
variable PBAR equal 1.0
velocity all create ${TK} 12345
fix TPSTAT all npt temp ${TK} ${TK} 100 iso ${PBAR} ${PBAR} 1000
Note que os valores de temperatura e pressão são especificados duas vezes. O primeiro valor é o valor do banho no início da simulação e o segundo o valor no final, sendo a temperatura e a pressão variadas linearmente entre esses dois valores ao longo da simulação. No caso, ambos os valores inicial e final são iguais, então, tanto a temperatura quanto a pressão do banho são mantidas constantes. Se quiséssemos fazer uma simulação variando a temperatura linearmente de 250 K para 400 K enquanto a pressão é mantida em 1 bar, poderíamos usar esse comando como:
fix TPSTAT all npt temp 250.0 400.0 100 iso 1.0 1.0 1000
As linhas a seguir controlam alguns outputs do programa. O comando thermo_style especifica os valores de quais variáveis devem ser impressas no arquivo de log gerado ao longo da simulação. São especificadas aqui, na sequência do comando: passo da simulação, tempo de CPU, energia total, energia cinética, energia de van der Waals, energia coulômbica devido a interações de curto alcance, energia coulombica devido a interações de longo alcance, temperatura, pressão, volume e densidade. Note que as variáveis evdwl, ecoul e elong não são incluídas pelo fftool, modifiquei o input gerado para incluí-las. Todas essas são variáveis já pré-definidas no LAMMPS, mas pode definir novas variáveis pelo comando variable e imprimi-las ao longo da simulação por meio do comando thermo_style. Para uma lista das variáveis pré-definidas, veja a página https://lammps.sandia.gov/doc/thermo_style.html. O comando seguinte define que tais variáveis devem ser calculadas e impressas a cada 100 passos de simulação (o padrão do fftool é a cada 1000, mas aumentei a frequência para ser impresso a cada 100).
O comando dump especifica como deve ser escrita a trajetória. O all indica que todas as partículas serão incluídas na trajetória, mas seria possível criar um grupo por meio do comando group do LAMMPS e incluir no arquivo trajetória apenas as partículas que corresponderem a esse grupo, isso pode ser útil por exemplo para gerar uma trajetória sem o solvente para poupar espaço caso nenhuma análise estrutural do mesmo interesse. 500 corresponde a frequência em que será escrita a trajetória (também alterei esse valor em relação ao padrão gerado pelo fftool, dump.lammpstrj é o nome do arquivo de trajetória gerado e as informações a seguir é o que será escrito para cada partícula no arquivo de trajetória: número do átomo, número da molécula, tipo do átomo, elemento, carga parcial, coordenadas cartesianas e em qual réplica da caixa a molécula está (útil para cálculo de propriedades dinâmicas. Note que alterei o padrão do fftool de x y z para xu yu zu, a diferença entre eles é que o primeiro “quebra” as moléculas ao passarem pelas bordas da caixa, colocando alguns átomos na outra face da caixa enquanto alguns continuam na original (mas lembre-se que essa quebra não é real, já que cada átomo interage com a réplica periódica mais próxima, apenas gera uma falha de visualização), enquanto o segundo registra o movimento contínuo da molécula, evitando essas aparentes “quebras” devido a condições periódicas de contorno. A escolha de um ou de outro não afetará em nada nenhuma análise feita, é apenas uma questão de preferência para visualização. O comando dump_modify atribui os elementos químicos a cada tipo atômico na sequência definida no arquivo data.lmp para registro na trajetória.
thermo_style custom step cpu etotal ke pe evdwl ecoul elong temp press vol density
thermo 100
dump TRAJ all custom 500 dump.lammpstrj id mol type element q xu yu zu ix iy iz
dump_modify TRAJ element C C H O H H O
A sequência a seguir, incluída pelo fftool, calcula o desvio quadrático médio dos átomos ao longo da simulação. Isso pode ser útil, por exemplo, para cálculos de coeficientes de difusão, mas não abordaremos isso nesse tutorial:
variable t equal time
compute MSD all msd com yes
variable msd equal c_MSD[4]
fix PRMSD all print 2000 “${t} ${msd}” file msd.lammps screen no
O próximo comando define uma variável vinst como sendo igual o volume instantâneo do sistema. A linha seguinte corresponde ao comando fix ave/time, que calcula o valor médio de uma variável ao longo de um determinado número de passos da simulação.
variable vinst equal vol
fix VAVG all ave/time 10 2500 25000 v_vinst
No comando fix ave/time são definidos 3 números que controlam a frequência de cálculos de média, especificados em sequência:
– Nevery: O valor da variável é calculado a cada Nevery (10) passos e somado para cálculo da média.
– Nrepeat: Número de passos a ser usado ao todo no cálculo de uma média (2500)
– Nfreq: Frequência em que a média será calculada (25000). Deve necessariamente ser um múltiplo dos dois anteriores.
No caso, usamos um total de 2500 valores tomados de 10 em 10 passos para o cálculo de uma média. Após 25000 passos, o cálculo é reiniciado. Se quiser simplesmente usar esse comando para calcular um valor uma vez a cada 50 passos, por exemplo, poderia usar fix (NOME) ave/time 1 1 50 (variável em questão).
A próxima linha comentada indicaria a frequência para escrever arquivos de restart, mas está comentada. No caso de cálculos mais longos é interessante usar essa opção para poder reiniciar em casos de quedas. A linha run indica o início da simulação com o número de passos. Alterei o padrão do fftool para fazer 200000 passos, como nosso passo de integração é 1 fs, isso corresponde a simular 200 ps no ensemble NPT.
#restart 10000 restart1.lmp restart2.lmp
run 200000
Os comandos a seguir são um exemplo interessante de uso de variáveis e médias não apenas como resultados a serem impressos, mas para alterar o próprio sistema modelo. Como em uma simulação NPT o volume continua flutuando mesmo após atingir o equilíbrio, se iniciássemos uma simulação NVT diretamente a partir da estrutura final, correríamos o risco de fazê-la com uma densidade um pouco diferente da densidade média do sistema. O ideal então é fazer a simulação NVT com o volume médio calculado após o sistema ter atingido o equilíbrio, e é justamente por isso que definimos o fix VAVG antes. Ao final da simulação, essa variável terá acumulado o volume médio dos últimos 25000 passos (note que para esse uso o número de passos deve ser um múltiplo de Nfreq, caso contrário resultará em um erro do tipo “fix not computed …”), e esse valor médio, provavelmente mais próximo ao valor de equilíbrio do que o volume instantâneo no último passo, será definido como o volume da simulação NVT. Para fazer a alteração de volume, define-se uma variável chamada lscale como a raiz cúbica da razão entre o volume médio calculado pelo fix ave/time e o volume instantâneo no último passo. O comando change_box vai escalar todas as dimensões da caixa multiplicando-as por lscale.
variable lscale equal (f_VAVG/v_vinst)^(1.0/3.0)
print “scaling coordinates by ${lscale}”
change_box all x scale ${lscale} y scale ${lscale} z scale ${lscale} remap
Os próximos comando desligam os comandos fix usados anteriormente para calcular o volume médio e o que definia o termostato e o barostato para o ensemble NPT para então podermos definir o termostato para a simulação NVT (não desativar um antes de ativar o outro resulta em erros). Faremos novamente 200000 passos (200 ps) de simulação. O último comando escreve um novo arquivo de estrutura do LAMMPS chamado data.eq.lmp, agora com as coordenadas finais. O mesmo poderá ser usado para iniciar novas simulações através dessa estrutura já equilibrada.
unfix VAVG
unfix TPSTAT
fix TSTAT all nvt temp ${TK} ${TK} 100
run 200000
write_data data.eq.lmp
.
.
5. Execução da simulação
Uma vez que os arquivos de input in.lmp e data.lmp estejam localizados no mesmo diretório, para executar a simulação, caso tenha instalado o LAMMPS diretamente de sua distribuição, basta executar um dos seguintes comandos (diferentes distribuições Linux e/ou versões do LAMMPS acabam sendo instaladas com nomes diferentes às vezes):
lammps -i in.lmp
lmp_daily -i in.lmp
Caso tenha compilado o binário ao invés disso, basta indicar o caminho e o nome dele aoinvés de lammps ou lmp_daily.
Opcionalmente, pode testar algum dos aceleradores de performance do LAMMPS (veja mais em https://lammps.sandia.gov/doc/Speed_packages.html) ou rodar em paralelo. O uso do acelerador OPT, por exemplo, sempre me dá um pequeno ganho de performance, para usá-lo, acrescente à linha de comando “-sf opt”. Com processadores e compiladores Intel, pode ser interessante a opção “-sf intel”. Caso vá usar sistematicamente o LAMMPS para alguma classe de problemas, vale a pena testar diferentes opções de otimização, mas, para efeito apenas de executar esse tutorial, isso não será necessário. Para executar em paralelo, tendo bibliotecas MPI configuradas em sua máquina, pode usar:
mpirun -np N lammps -i in.lmp
ou
mpirun -np N lmp_daily -i in.lmp
onde N é o número de nós a serem usados.
Após começar a execução, diversas informações começarão a aparecer na tela, incluindo os comandos presentes no arquivo in.lmp. Ao começar de fato a simulação, as informações específicadas pelo comando thermo_style no arquivo in.lmp serão exibidas com a frequência de passos definida no comando thermo. Deverá ver algo similar ao mostrado abaixo:
Step CPU TotEng KinEng PotEng E_vdwl E_coul E_long Temp Press Volume Density
0 0 1729.4053 1877.0161 -147.61084 -787.9711 9387.2516 -9089.0216 300 20014.884 32768 0.70037263
100 3.9124149 1441.9678 1753.2016 -311.23383 -988.83127 8978.5972 -9149.8798 280.21096 -4933.4493 33234.686 0.6905379
200 7.80679 1565.8549 1816.5681 -250.71319 -955.94078 8861.6135 -9163.7562 290.33871 6941.3768 33965.972 0.67567064
300 11.684727 1630.1034 1837.1221 -207.0186 -847.5157 8734.4815 -9165.2215 293.62381 -4141.7082 34658.963 0.6621609
400 15.821211 1598.4492 1940.1294 -341.6802 -847.76126 8692.7518 -9175.0318 310.08728 5207.1449 35209.881 0.65180029
Isso permite acompanhar o andamento da simulação, lembrando que serão feitas duas etapas, sendo a primeira a pressão constante e a segunda a volume constante. Essas mesmas informações serão escritas no arquivo lammps.log. Veremos como extrair informações dele na próxima página.
.
.
6. Visualização da trajetória
Após a conclusão da trajetória, poderá visualizá-la utilizando o VMD ou algum outro programa de visualização compatível. Para isso use o comando:
vmd dump.lammpstrj
Você pode acrescentar as arestas da caixa de simulação pelo menu “Extensions” > “TKconsole” no VMD e entrar com o comando:
pbc box
Também pode alterar as representações pelo menu “Graphics” > “Representations”. Na imagem abaixo é mostrada uma estrutura da simulação com a representação “VDW”.
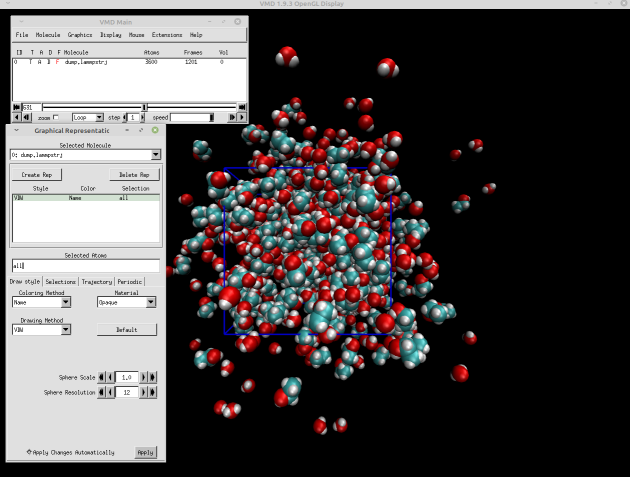
Figura 3 – Representação de van der Waals (VDW) da caixa de simulação no VMD.
Note que logo após a primeira frame, moléculas começarão a aparentemente sair da caixa de simulação. Isso deve-se a opção “xu yu zu” usada no comando dump, que faz com que seja registrado o movimento contínuo das moléculas, sem “saltos” para a metade oposta ao passar por uma das bordas da caixa. Entretanto, conforme descrito anteriormente, cada molécula está o tempo todo rodeada de outras moléculas de água e etanol devido às condições periódicas de contorno e, após alguma variação no início da simulação, o volume passa a apenas flutuar em torno de um valor médio. Pode notar isso pelas arestas plotadas junto com as estruturas, o que permite ver que, mesmo que as moléculas pareçam se afastar, a densidade do sistema permanece aproximadamente constante. Pode acrescentar réplicas periódicas na visualização através da aba “Periodic” na janela “Graphic Representations” do VMD para uma melhor visualização desse efeito.
Se ao invés de “xu yu zu” usasse “x y z” no comando dump do arquivo in.lmp, cada átomo iria desaparecer e reaparecer na face oposta da caixa ao passar por suas extremidades. Essa visualização teria a vantagem de mostrar de modo mais claro a densidade do sistema, mas as moléculas apareceriam frequentemente “quebradas”. Para as análises que faremos na próxima parte desse tutorial, tal escolha é irrelevante, pode escolher a forma de gerar a trajetória que achar mais conveniente.


Pingback: Tutorial LAMMPS – água e etanol | kalilbn
Está rodando, tutorial funcionou perfeitamente! Obrigado pela didática e sugestões! Aguardando as próximas partes!
Pingback: Script em python para extrair dados dos arquivos de log do LAMMPS | kalilbn
Pingback: Nova versão do programa para extrair dados do arquivo de log do LAMMPS | kalilbn