Tutorial Gromacs – água pura – parte 2
Enquanto na página anterior descrevemos como instalar os programas necessários, preparar a caixa de simulação e executar a minimização de energia e a simulação de dinâmica molecular, nessa página focaremos na realização de análises através dos outputs gerados. Começaremos com a análise de componentes de energia e outras funções de estado através do arquivo ener.edr gerado. Em seguida, realizaremos análises estruturais, incluindo análises de ligações de hidrogênio e distribuição radial de pares usando ferramentas do próprio Gromacs. Finalmente, repetiremos as análises de distribuição radial de pares, porém usando outro programa de análise chamado Travis e o usaremos para calcular distribuições espaciais (sdf).
.
Conteúdos:
Página 1 (ir para a página)
- Instalação dos programas utilizados
- Criação e visualização da caixa de simulação
- Minimização de energia
- Execução da simulação
- Receita de bolo (tutorial sem explicações)
.
Página 2
- Componentes de energia, densidade, volume, temperatura e pressão
- Ligações de hidrogênio
- Distribuição radial de pares com o Gromacs
- Distribuição radial de pares e distribuição espacial com o Travis
- Problemas propostos
- Referências
.
1. Componentes de energia, densidade, volume, temperatura e pressão
Componentes de energia e diversas outras grandezas físicas são calculadas ao longo da simulação e armazenadas no arquivo ener.edr. Para extrair essas informações, usamos o programa gmx energy, que já usamos na página anterior para analisar a variação da energia potencial na minimização de energia. Começaremos pela energia potencial. Assim como na página anterior, execute o comando
gmx energy -f ener.edr -o potential
Escolha o número correspondente a opção “Potential” e dê Enter 2 vezes. Abra o gráfico gerado com o xmgrace:
xmgrace potential.xvg

Note que o perfil de energia potencial em uma simulação a temperatura constante é bem diferente do observado durante a minimização de energia, onde notamos um decréscimo contínuo da energia potencial. Em uma simulação a temperatura constante a energia potencial pode tanto aumentar (como ocorreu nesse caso) ou diminuir até o sistema atingir o estado de equilíbrio, no qual a energia potencial (e todas as outras componentes de energia) vão apenas oscilar em torno de um valor médio. A figura abaixo mostra a variação nos primeiros picossegundos de simulação.
A energia potencial calculada, bem como qualquer outra componente de energia calculada pelo gmx energy, é uma propriedade extensiva, ou seja, depende do tamanho do sistema. Um sistema com o dobro de moléculas apresentaria (exceto por possíveis artefatos de tamanho pequeno) o dobro da energia potencial. Podemos transformá-la em uma propriedade intensiva dividindo pelo volume (o que resultaria em uma densidade de energia potencial) ou pelo número de moléculas. Isso pode ser feito no xmgrace no menu “data” > “transformations” > “geometric transform”. Na janela que abrirá, altere o campo “Scale Y” por 1/1500 para dividir a energia potencial ponto a ponto pelo número de moléculas na caixa. Dê “Apply” e feche essa janela. Clique no botão “Auto scale” (símbolo AS) no lado esquerdo do gráfico para rescalar automaticamente. O gráfico de energia potencial agora é a energia potencial por mol de água, sendo uma propriedade intensiva.
De modo similar, podemos gerar o gráfico da densidade em função do tempo:
gmx energy -f ener.edr -o density
Escolha a opção “density” e abra o gráfico no xmgrace:
xmgrace density.xvg
Há um rápido aumento de densidade no início da simulação e, assim como a energia potencial, após essa relaxação inicial a densidade oscila em torno de um valor médio de aproximadamente 970 kg/m³. Podemos filtrar a flutuação de alta frequência do gráfico calculando a média móvel no xmgrace. Para isso, vá em “Data” > “Transformations” > “Running Averages”. Na nova janela, escolha a cada quantos pontos tomará a média móvel . Fazendo a cada 500 pontos, temos um resultado similar ao mostrado abaixo, onde a curva preta é o dado bruto e a vermelha a média móvel:

Gere e visualize as variações da temperatura e da pressão usando o gmx energy de modo similar e trocando o nome do output na opção -o. Observe que enquanto a temperatura, a densidade e a energia apresentam flutuações relativamente pequenas após o sistema relaxar, a pressão apresenta flutuações gigantescas. Isso ocorre pois em um sistema muito pequeno flutuações mínimas no volume são suficientes para gerar efeitos bastante significativos sobre a pressão.
Podemos quantificar o valor médio e o desvio-padrão dessas propriedades após o sistema atingir o equilíbrio usando o programa do gromacs gmx analyze, onde com a opção -f é indicado o arquivo de input, que deve ser um dos arquivos xvg gerados anteriormente, e com a opção -b o tempo no qual a análise deverá ser iniciada. Vamos descartar os primeiros 50 ps. Para a densidade, por exemplo, execute:
gmx analyze -f density.xvg -b 50
Na tabela abaixo são dados os valores médios e respectivos desvios das propriedades calculadas. Note que, apesar de termos definido a pressão externa como 1,0 bar, a pressão média foi 2,8 bar. Isso torna difícil avaliar efeitos de pequenas variações de pressão em simulações de dinâmica molecular, mas, felizmente, a menos que a pressão varie em ordens de grandeza os efeitos sobre as propriedades de líquidos são normalmente negligenciáveis.
Tabela 1 – Valor médio e desvio-padrão de propriedades selecionadas na simulação de água após o sistema atingir o equilíbrio
| Propriedade | Média | Desvio-padrão |
| Energia potencial (kJ/mol) | -63974 | 323 |
| Densidade (kg/m³) | 1007,2 | 5,4 |
| Temperatura (K) | 299,9 | 3,8 |
| Pressão (bar) | 5 | 459 |
.
2. Ligações de hidrogênio
Iniciaremos agora as análises estruturais utilizando a trajetória gerada. Primeiramente, vamos analisar as ligações de hidrogênio formadas usando o programa gmx hbond. Vamos considerar aqui o número de ligações de hidrogênio formadas e as distribuições de distância e ângulo das mesmas. Essas três análises são feitas simultaneamente usando o comando:
gmx hbond -f traj_comp.xtc -b 50 -num hbnum -dist hbdist -ang hbang
A opção -b indica para pular os primeiros 50 ps para a relaxação do sistema. A opção -num gera um gráfico do número de ligações de hidrogênio ao longo tempo, -dist dá a distribuição de comprimento das ligações e -ang a distribuição de ângulo hidrogênio – doador – aceptor. Após executar o comando será pedido para escolher 2 tipos de moléculas. No caso só temos moléculas de água, então selecione as duas vezes a opção “SOL”. Em uma mistura de 2 ou mais espécies capazes de realizar ligações de hidrogênio poderíamos analisar as ligações entre cada par possível separadamente.
Começaremos analisando o gráfico hbnum.xvg. Esse gráfico possui duas curvas: Além do número de ligações de hidrogênio, também possui uma curva com o número de pares aceptor – doador em uma distância de até 0,35 nm que poderiam em princípio formar uma ligação de hidrogênio. Para abrir as duas curvas simultaneamente no xmgrace, use a opção -nxy:
xmgrace -nxy hbnum.xvg
Ambas as curvas flutuam em torno de um valor médio no sistema em equilíbrio. Podemos obter os valores médios e os respectivos desvios usando o comando gmx analyze como feito anteriormente:
gmx analyze -f hbnum.xvg
Os resultados da primeira curva (número de ligações de hidrogênio) aparecerão na primeira linha enquanto os resultados da segunda curva (número de pares) aparecerão na segunda linha. Note que esses valores são para todo o sistema, podemos dividi-los pelo número de moléculas para termos o número de ligações de hidrogênio por molécula. Na tabela abaixo são mostrados os valores obtidos.
Tabela 2 – Número de ligações de hidrogênio e de pares de moléculas de água em distâncias de até 0,35 nm.
| Por todo o sistema | Por molécula | |||
| Propriedade | média | desvio-padrão | média | desvio-padrão |
| N⁰ de ligações | 2724 | 16 | 1,82 | 0,01 |
| N⁰ de pares | 4940 | 69 | 3,29 | 0,05 |
Note que os números de ligações por molécula na Tabela 2 é na verdade metade do número de ligações que uma molécula está realizando com as demais, pois se a molécula A está realizando uma ligação de hidrogênio com a molécula B, essa ligação não deve ser contabilizada novamente ao observarmos que B também está realizando a mesma ligação de hidrogênio com A. Dessa forma, cada molécula de água está realizando na média 2×1,82 = 3,64 ligações de hidrogênio com outras moléculas, mas contabilizamos 1,82 para evitar contagem dupla. O valor de 3,64 é um pouco menor do que a estrutura idealizada do gelo, onde cada molécula de água realiza 4 ligações (2 recebendo e 2 doando) ligações de hidrogênio com suas vizinhas.
Visualize agora as distribuições de comprimento e ângulo de ligações de hidrogênio abrindo os gráficos hbdist.xvg e hbang.xvg no xmgrace. Na figura abaixo são mostrados os resultados obtidos. Note que pelos critérios do programa só se considera a formação de uma ligação de hidrogênio para distância aceptor-doador de até 0,35 nm e ângulo hidrogênio-doador-aceptor de até 30⁰, apesar que esses valores podem ser ajustados usando as opções -r e -a, respectivamente.

A distribuição de distância aceptor-doador (ou seja, entre os átomos de O) para moléculas de água realizando ligações de hidrogênio é relativamente estreita, com máximo de probabilidade em 0,28 nm. Essa distância será importante também nas análises de distribuição radial de pares como veremos a seguir. A distribuição do ângulo hidrogênio-doador-aceptor, por outro lado, é bastante larga, com máximo em 10⁰, indicando que os átomos envolvidos na ligação de hidrogênio tendem a estarem quase colineares. Na verdade, o ângulo de 0⁰ corresponderia a menor energia, como seria observado fazendo uma otimização de geometria de um par de moléculas de água formando ligação de hidrogênio, mas por razões entrópicas o ângulo de 0⁰ é desfavorável e o máximo ocorre em um ângulo ligeiramente maior. A figura abaixo mostra um par de moléculas ligadas por ligação de hidrogênio com valores de distância e ângulo próximas aos máximos das distribuições, com distância aceptor-doador de 0.28 nm e ângulo hidrogênio-doador-aceptor de 8⁰:

.
3. Distribuição radial de pares com o Gromacs
Enquanto na seção anterior tratamos de análises específicas para moléculas capazes de realizar ligações de hidrogênio, a distribuição radial de pares tem caráter mais geral e pode ser usada em qualquer sistema. Denotada como g(r) ou rdf (do inglês “radial distribution function”), a distribuição radial de um átomo, molécula ou estrutura B ao redor de um átomo, molécula ou estrutura A de referência é definida como:

onde rA é a distância em relação ao sítio de referência A, ρB(rA) é a densidade da espécie B em uma casca esférica de raio rA e espessura dr ao redor da espécie de referência A e ρB,sistema é a densidade média de B em todo o sistema (isso é, número total da espécie B dividida pelo volume da caixa de simulação). Dessa forma, a distribuição radial de pares é uma densidade normalizada em função da distância de uma espécie de referência. Para um sistema sem nenhuma interação, ou seja, um gás ideal, teríamos g(r) = 1 para qualquer r, pois a densidade ao redor da espécie de referência seria uniforme. g(r) > 1 indica uma densidade maior do que em um sistema sem interações com o mesmo volume e mesmo número de partículas, sendo portanto uma região termodinamicamente favorável para aquela espécie. g(r) < 1 indica uma região com densidade na distância r menor que o esperado para um sistema sem interações, indicando portanto uma região desfavorável do ponto de vista termodinâmico.
Para realizar a análise da distribuição radial de pares com o Gromacs iremos primeiramente criar um arquivo de índice indicando átomos de interesse para a análise. Para isso, execute o comando:
gmx make_ndx -f conf.gro
em seguida, digite:
a OW
a HW*
q
A linha “a OW” cria um grupo com todos os átomos com nome OW (nome dado aos átomos de oxigênio da água no conf.gro) já a linha “a HW*” cria outro grupo com os átomos de hidrogênio (os nomes desses no conf.gro aparecem como HW1 e HW2, usar “HW*” indica que estamos selecionando ambos). Será criado o arquivo index.ndx, que pode ser visualizado com um editor de textos como o vi. Esse arquivo contém o conjunto de índices de átomos em cada grupo definido, incluindo alguns padrão do gromacs e os que acabamos de definir: O grupo OW contendo os índices de todos os átomos de oxigênio e o HW contendo os indices de todos os átomos de hidrogênio.
Para calcularmos a distribuição radial de pares usaremos o programa gmx rdf lendo o arquivo index.ndx gerado e a trajetória traj_comp.xtc. Para calcular a distribuição entre átomos de oxigênio, use o comando:
gmx rdf -f traj_comp.xtc -b 50 -bin 0.01 -n index.ndx -o rdf-Ow-Ow -cn cn-Ow-Ow
e para as seleções escolha 2 vezes o grupo OW, dando Enter entre eles. Em seguida, aperte Ctrl+D para finalizar. A opção -b 50, assim como vimos anteriormente, é usada para ignorar os primeiros 50 ps nas análises e opção -bin determina a espessura em nm das cascas esféricas consideradas no cálculo da distribuição radial. Um valor muito pequeno leva a gráficos ruidosos já um valor muito grande leva a perda de resolução. A distribuição radial de pares pode ser vista abrindo o gráfico rdf-Ow-Ow.xvg no xmgrace:
xmgrace rdf-Ow-Ow.xvg
Esse gráfico é mostrado na figura abaixo. Em distâncias muito curtas temos g(r) = 0, indicando a ausência de átomos de oxigênio nessas distâncias de um átomo de referência. Essa região corresponde ao volume excluído devido a região fortemente repulsiva do potencial de Lennard-Jones que impede um átomo de “entrar dentro” do outro. Após isso temos um pico intenso com máximo em 0,28 nm, a mesma distância que vimos anteriormente como máximo da distribuição aceptor – doador para moléculas de água formando ligações de hidrogênio. Esse primeiro máximo corresponde a primeira camada de coordenação, ou seja, aos primeiros vizinhos da molécula de referência, e costuma ser o mais intenso para líquidos puros e em soluções homogêneas, sendo particularmente intenso e estreito para espécies capazes de formar ligações de hidrogênio. Após esse primeiro máximo, temos um mínimo local com g(r) < 1 seguido por um novo máximo local, porém mais alargado e menos intenso que o primeiro. As posições de mínimos em líquidos simples são interpretadas como as regiões entre camadas de coordenação, desse modo, o primeiro mínimo marca o fim da primeira camada e o segundo máximo corresponde a segunda camada de coordenação.
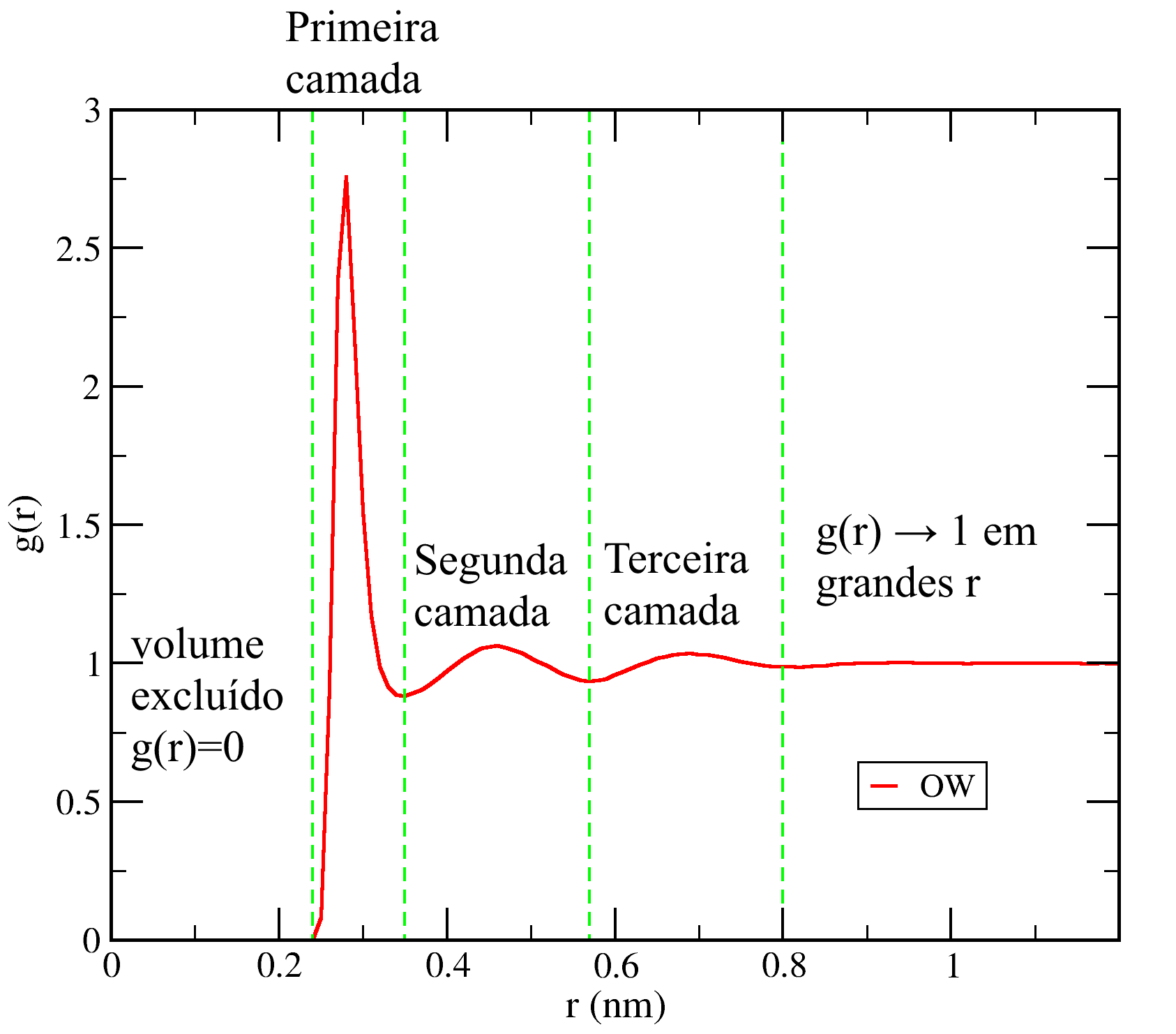
As amplitudes dos máximos e mínimos seguintes vão se tornando cada vez menos intensas, mostrando que a influência da molécula de referência sobre a organização das demais moléculas diminui rapidamente a medida que consideramos camadas cada vez mais distantes. Tal resultado corrobora a afirmação tradicional de que os líquidos apresentam apenas organização de curto alcance. Resultados bem diferentes são observados para sólidos cristalinos, vide essa página.
Além do gráfico de distribuição radial de pares, foi gerado também o gráfico do número cumulativo no arquivo cn-OW-OW.xvg, que se relaciona com a integral da função g(r):
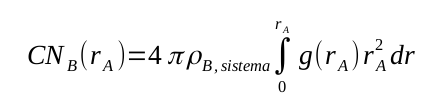
onde o termo 4π surge das integrais sobre coordenadas angulares em um sistema de coordenadas esféricas. O número cumulativo na posição rA dá o número médio de partículas da espécie B que estão até a uma distância rA da espécie A de referência. Como dito anteriormente, interpretamos a região até o primeiro mínimo local da função g(r) como sendo a primeira camada de coordenação. Uma pergunta interessante seria então quantas moléculas de água estão na primeira camada, ou, de modo equivalente, quantos vizinhos uma molécula de água possui em média? Podemos responder a isso abrindo o arquivo cn-Ow-Ow.xvg ou no xmgrace ou mesmo em um editor de texto e buscando o valor do número cumulativo na posição do primeiro mínimo da distribuição entre os átomos de oxigênio. No caso, o primeiro mínimo ocorre em 0,35 nm e o número cumulativo nessa posição é 5,09. Podemos dizer então que há em média 5,09 moléculas de água na primeira camada de cada molécula de água. Isso é mais do que o valor de 4 vizinhas esperada pela estrutura do gelo, onde cada molécula faz ligações de hidrogênio com outras 4. O fato da água acomodar mais moléculas na primeira camada do que o gelo explica o comportamento anômalo dela apresentar maior densidade na fase líquida do que na fase sólida.
Para determinarmos a quantidade de moléculas na segunda camada olhamos para o valor do número cumulativo na posição do segundo mínimo de g(r) (0,57 nm), obtendo o valor de 24,08. Esse valor, entretanto, corresponde às duas primeiras camadas em conjunto, para saber quantas temos na segunda camada subtraímos o valor anterior de moléculas na primeira camada e chegamos em aproximadamente 19 moléculas de água na segunda camada.
Além da distribuição entre átomos de oxigênio, podemos analisar também a distribuição de átomos de hidrogênio ao redor dos átomos de oxigênio, com o comando:
gmx rdf -f traj_comp.xtc -b 50 -bin 0.01 -n index.ndx -o rdf-Ow-Hw -cn cn-Ow-Hw
e escolhendo agora os grupos “OW” e “HW*”. Ao abrir a distribuição radial resultante no xmgrace, note um pico muito intenso e estreito em 0,1 nm. Se observar o número cumulativo correspondente a esse pico, verá que corresponde a exatamente 2 átomos de hidrogênio. Esse primeiro pico corresponde aos átomos de hidrogênio na mesma molécula do átomo de oxigênio de referência, enquanto apenas do segundo pico em diante correspondem aos átomos de hidrogênio das outras moléculas de água. Normalmente estamos interessados em observar a estrutura intermolecular separadamente da intra-molecular, para isso podemos excluir os átomos que fazem parte da molécula de referência na análise adicionando as opções -excl e -s topol.tpr na linha de comando:
gmx rdf -f traj_comp.xtc -b 50 -bin 0.01 -n index.ndx -o rdf-Ow-Hw -cn cn-Ow-Hw -excl -s topol.tpr
O novo gráfico rdf-Ow-Hw.xvg não inclui mais os átomos de hidrogênio da molécula de referência. Podemos visualizá-lo junto com o gráfico da distribuição entre átomos de oxigênio usando o comando:
xmgrace rdf-Ow-Hw.xvg rdf-Ow-Ow.xvg -legend load
onde a opção “-legend load” carrega os nomes dos respectivos arquivos como legendas. O gráfico resultante é mostrado na figura abaixo, onde a curva preta é a distribuição de átomos de hidrogênio e a vermelha a distribuição de átomos de oxigênio em torno de um átomo de oxigênio de referência:

A distribuição radial de pares de hidrogênio ao redor de oxigênio apresenta dois picos particularmente intensos. O primeiro, com máximo em 0,18 nm, corresponde a átomos de hidrogênio que estão formando ligações de hidrogênio com o oxigênio de referência, sendo esse valor de 0,18 nm típico para a distância aceptor-hidrogênio em ligações de hidrogênio onde o aceptor é o oxigênio. Ao olharmos o número cumulativo no gráfico cn-Ow-Hw.xvg até a posição do primeiro mínimo dessa distribuição (0,24 nm), encontramos o valor de 1,8 átomos de hidrogênio envolvidos nas ligações de hidrogênio que a molécula de referência recebe, o que é apenas um pouco menor que o valor de 2,0 que seria esperado na estrutura idealizada do gelo. O segundo máximo da distribuição O-H ocorre um pouco após o primeiro máximo da distribuição O-O e corresponde ao mesmo tempo aos átomos de hidrogênio das moléculas na primeira camada que não estão fazendo ligações de hidrogênio com a molécula de referência e também inclui átomos de hidrogênio da segunda camada de coordenação.
Apesar da distribuição radial de pares poder ser calculada também para sistemas onde há múltiplas fases, a interpretação dos resultados demanda mais cuidado pois dependendo da distância r podemos amostrar fases distintas. Esse ponto será trabalhado em um dos problemas propostos ao final desse tutorial.
.
4. Distribuição radial de pares e distribuição espacial com o Travis
Aqui realizaremos análises de distribuição radial de pares similares às feitas anteriormente com o Gromacs mas agora usando o programa Travis. Apesar de que vamos realizar as mesmas análises, será uma introdução útil ao programa Travis que permite realizar, além de diversas outras análises, cálculo de distribuições radiais com condições mais sofisticadas que o Gromacs.
O Travis não trabalha diretamente com trajetórias em formato xtc, temos que antes de mais nada convertê-la em um formato que ele leia, para isso vamos convertê-la para o formato pdb e descartar os primeiros 100 ps de simulação usando o comando:
gmx trjconv -f traj_comp.xtc -b 100 -pbc mol -o traj.pdb
Ao ser perguntado sobre quais grupos salvar, escolha todo o sistema (opção 0). Agora leia a trajetória resultante com o travis usando:
travis traj.pdb
O travis funciona de modo interativo, te fazendo perguntas sobre opções que deseja usar no próprio terminal. Ao perguntar sobre atribuir tipos atômicos aos átomos Hw e Ow responda “y” e ao perguntar sobre se quer fazer isso automaticamente responda “y” de novo.
Veja que eles foram identificados corretamente como hidrogênio e oxigênio.
Ao perguntar sobre usar o modo avançado escolha “n” para não e escolha “y” para confirmar as dimensões encontradas para a caixa e que é uma caixa com condições periódicas de contorno.
O Travis tenta identificar as moléculas com base em distâncias interatômicas, diferente dos programas de análise do Gromacs que usam a topologia para tal, portanto, é importante verificar nessa etapa se a identificação foi feita de modo correto. No meu caso, ele identificou quatr tipos de moléculas (H2O, HO, H e O) enquanto deveria ter identificado apenas moléculas de H2O, como mostrado abaixo:
*** Molecule 1: H2O; Representant 1; Distances in pm ***
H1 H2 O1
H1 *** – 101
H2 – *** 101
O1 101 101 ***
Atoms H1, H2 are equivalent.
*** Molecule 2: HO; Representant 1; Distances in pm ***
H1 O1
H1 *** 100
O1 100 ***
*** Molecule 3: O; Representant 1; Distances in pm ***
[only 1 Atom]
*** Molecule 4: H; Representant 1; Distances in pm ***
[only 1 Atom]
*** The following 4 kinds of molecules have been recognized:
(ordered by molecular mass)
– Molecule 1: H2O (1388 pieces, 18.02 g/mol)
(2 noneq. atoms, 1 noneq. bond, 1 noneq. angle)
– Molecule 2: HO (111 pieces, 17.01 g/mol)
(2 noneq. atoms, 1 noneq. bond, 0 noneq. angles)
– Molecule 3: O (1 piece, 16.00 g/mol)
(1 noneq. atom, 0 noneq. bonds, 0 noneq. angles)
– Molecule 4: H (113 pieces, 1.01 g/mol)
(1 noneq. atom, 0 noneq. bonds, 0 noneq. angles)
Isso indica que precisamos alterar o critério de distância para evitar que enxergue essas moléculas de água como “quebradas”. Responda portanto “n” em ambas as perguntas:
“Create images of the structural formulas (y/n)?”
“Accept these molecules (y) or change something (n)? [yes]”
Ele dará 3 opções para alterar as moléculas, escolha a “1” (“Change covalent atom radii used for bond recognition”)
Agora escolha o átomo Ow e coloque o raio como 70.0 e depois o Hw e coloque 42.5.
Feito isso, ao perguntar novamente qual átomo quer alterar, digite “Enter” para voltar e depois Enter novamente ao mostrar o menu de alterações.
Se tudo der certo, ele deve identificar um único tipo de molécula:
*** Molecule 1: H2O; Representant 1; Distances in pm ***
H1 H2 O1
H1 *** – 101
H2 – *** 101
O1 101 101 ***
Atoms H1, H2 are equivalent.
*** The following 1 kind of molecules has been recognized:
(ordered by molecular mass)
– Molecule 1: H2O (1500 pieces, 18.02 g/mol)
(2 noneq. atoms, 1 noneq. bond, 1 noneq. angle)
Ao perguntar novamente “Create images of the structural formulas (y/n)? [no] ” responda “n” e, depois, ao perguntar se aceita as moléculas, responda “y”.
Ele vai dar agora o menu com as propriedades que pode calcular.
Vamos fazer primeiro a distribuição radial de pares. Digite “rdf” e dê Enter.
Na pergunta sobre usar o modo avançado responda “n”. Sobre fazer o cálculo intermolecular ou intramolecular, escolha intermolecular (opção 1).
Como só há um tipo de molécula, a escolha sobre qual molécula será usada como referência é irrelevante. Vamos escolher “0” e “1”.
Sobre qual átomo selecionar, escolha o oxigênio nas duas vezes (“O”).
Para todas as demais perguntas, usaremos a opção padrão então basta dar “Enter” até ele iniciar os cálculos. Poderíamos na pergunta “In which time step to start processing the trajectory? ” escolher um número de passos a serem pulados do início da trajetória para garantir a relaxação (similar a opção -b do Gromacs, mas note que aqui é em número de estruturas salvas, não em tempo), mas, como já descartamos o início da trajetória para gerar o pdb lido pelo travis, usaremos em todas as análises aqui a trajetória completa. Será gerado o arquivo rdf_H2O_#2_H2O_[Or_Oo].agr que pode ser visualizado com o xmgrace:
xmgrace rdf_H2O_#2_H2O_[Or_Oo].agr
Note que enquanto o Gromacs trabalha com a distância r em nm o Travis trabalha em pm!
Para calcularmos a distribuição de átomos de hidrogênio ao redor do oxigênio, poderíamos repetir todo o passo a passo anterior. Mas felizmente o Travis possui também uma opção mais ágil. Ele gera um arquivo de texto com nome input.txt com todas as opções que selecionamos, então podemos simplesmente editar esse arquivo com o que queremos modificar na nova análise e manter o restante igual. Vamos primeiro copiá-lo para um novo arquivo input_rdf.txt:
cp input.txt input_rdf.txt
Abra o arquivo input_rdf.txt em um editor de textos e na pergunta “! Which atom(s) to take from OM H2O (e.g. “C1,C3-5,H”, “*”=all)? [#2]”, altere a resposta na linha abaixo dela de O para H. Agora execute o travis lendo esse arquivo de input e a trajetória pdb com o comando:
travis -i input_rdf.txt -p traj.pdb
Para o cálculo da distribuição espacial, edite o arquivo de input e delete a linha onde foi escolhida a opção “rdf” e todas as linhas abaixo dela. Execute novamente o programa travis e escolha a opção “sdf”.
Para os átomos de referência, escolha os 3 únicos átomos da molécula de água digitando O,H1,H2. Em seguida, selecione a opção intermolecular “1”. Para os átomos a serem observados, escolha o oxigênio da água, “O”. Para a quantidade de bins, aumentaremos para 150 ao invés do padrão de 100.
Para visualizar o sdf, abra primeiro a estrutura de referência gerada no VMD com o comando
vmd ref_h2o_o1_h1_h2.xyz
Esse arquivo de estrutura contém uma única molécula de água. Antes de avançarmos, vá em “Graphics” > “Representations” e mude a representação da molécula para o modelo CPK. Agora, vamos abrir a distribuição espacial. Clique com o botão direito na área branca da janela VMD main e escolha a opção “new molecule” e, na nova janela, clique no botão “Browse…”. Carregue o arquivo sdf_H2O_O1H1H2_O.s2.cube e clique em Load. Inicialmente não aparecerá nada apesar do arquivo ter sido carregado. É necessário gerar uma representação para a distribuição espacial. Para isso, clique em “Graphics” > “Representations”. No topo da janela de representações, onde aparece “Selected Molecule”, verifique que está selecionado o arquivo de sdf. Mude a opção “Drawing Method” para “Isosurface”. Aparecerão outra opções na mesma janela. Altere a opção “Draw” para “Solid Surface” ou para “wireframe”. Em seguida, ajuste o isovalor no campo “isovalue” ou digitando ou movendo a barra ao lado dele. Observe como a superfície de sdf muda. A superfície mostrada corresponde aos pontos ao redor da molécula de referência onde a densidade média de átomos de oxigênio por nm³ é igual ao isovalor selecionado. Dessa forma, quanto maior o isovalor, menores tendem a ser as superfícies mostradas. O valor de 85 nm⁻³ mostra apenas as regiões de maior densidade na primeira camada, que é a mais estruturada, e vemos 4 regiões de máxima densidade que correspondem a descrição da estrutura tetraédrica da água líquida, onda cada molécula participa de ligações de hidrogênio com 4 vizinhas, sendo as duas regiões de máximos alinhadas com as ligações O-H correspondentes a moléculas que recebem ligação de hidrogênio da molécula de referência e as duas outras regiões de máximo correspondem a moléculas que doam ligações de hidrogênio para a de referência. Você pode alterar as cores das superfícies na janela de “Graphical representations” escolhendo o “Coloring method” como “Color ID” e escolhendo a cor no campo que aparecerá ao lado desse. Escolhi a cor vermelha para a distribuição de oxigênio. O resultado com isovalor 85 nm-3 é mostrado abaixo:

Valores menores mostrarão outras regiões da primeira camada onde há probabilidades significativas porém menores de encontrar outra molécula de água. Lembre-se da análise de número cumulativo que foi discutida anteriormente, há na média mais do que 4 vizinhas na primeira camada, de modo que o líquido não segue exatamente a estrutura tetraédrica esperada no gelo apesar de as regiões mais favoráveis para encontrar outras moléculas de água ainda serem nos vértices do tetraedro ao redor da molécula de referência.
Faça a mesma análise para os átomos de hidrogênio. Pode, assim como no caso da distribuição radial, copiar o input.txt gerado, alterar o átomo a ser analisado e rodar o travis lendo o arquivo de input junto com a trajetória. Pode carregar as duas distribuições ao mesmo tempo no VMD. Abaixo são mostradas as distribuições espaciais do oxigênio em vermelho e do hidrogênio em cinza, sendo as superfícies sólidas criadas com isovalor maior e as em wireframe com isovalor menor (clique na imagem para ver a rotação da distribuição caso não reproduza automaticamente):

.
5. Problemas propostos:
Realize uma simulação de uma interface água vapor fazendo as seguintes etapas:
a) Copie o arquivo .gro gerado pelo gmx solvate para um novo diretório com o nome conf.gro (não copie a estrutura minimizada nem o confout.gro da simulação, use a estrutura gerada pelo gmx solvate!)
b) Edite o arquivo conf.gro em um editor de textos e, na última linha, onde temos as dimensões da caixa de simulação, altere o valor da dimensão z para 10.0 nm. Visualize esse arquivo no vmd colocando as arestas da caixa. Por causa das condições periódicas de contorno, são geradas na verdade 2 interfaces água/vapor. Uma em z = 0 e outra em z ~ 3,7 nm.
c) Copie os arquivos topol.top e grompp_minimize.mdp e execute a minimização de energia exatamente como no tutorial.
d)Para executar a simulação, copie o arquivo confout.gro resultante da etapa 3 e o renomeie para conf.gro. Copie também para o mesmo diretório os arquivos topol.top e grompp.mdp usados na simulação.
e)Para a simulação das interfaces, vamos desligar o acoplamento de pressão. Para isso, edite o arquivo grompp.mdp e na linha “pcoupl =” coloque “no” após o sinal de igual. Além disso, apague as linhas iniciadas com “ref_p”, “compressibility”, “tau_p” e “pcoupltype”.
f) Execute a simulação usando os comandos “gmx grompp” e “gmx mdrun”
g) Realize as análises de ligações de hidrogênio e distribuição radial de pares como feitas no tutorial. Como o número médio de ligações de hidrogênio é afetado pela presença das interfaces? Compare os perfis de distribuição radial de pares, note que no sistema interfacial o mesmo não converge para 1.0, mas passa a apresentar um decréscimo contínuo. Por que isso ocorre?
.
6. Referências
Softwares utilizados:
- van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E. & Berendsen, H. J. C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 26, 1701 (2005).
- Abraham, M. J., Murtola, T., Schulz, R., Pálla, S., Smith, J. C., Hess, B. & Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19 (2015).
- Humphrey, W., Dalke, A., Schulten, K. “VMD – Visual molecular dynamics” J. Mol. Graphics 14.1, 33–38 (1996).
- Brehm, M., Kirchner, B. “TRAVIS—A free analyzer and visualizer for Monte Carlo and molecular dynamics trajectories” J. Chem. Inf. Model. 51, 2007–2023 (2011).
Algoritmos e campos de força:
- Bussi, G., Donadio, D., Parrinello, M. “Canonical sampling through velocity rescaling,” J. Chem. Phys., 126 014101 (2007).
- Darden, T., York, D., Pedersen, L. “Particle mesh Ewald: An N∙log(N) method for Ewald sums in large systems,” J. Chem. Phys., 98 10089–10092 (1993).
- Berendsen, H. J. C.; Postma, J. P. M.; van Gusteren, W. F. Hermans, J. In Intermolecular Forces; Pullman, B., Ed.; Reidel: Dordrecht, 1981.
.
.
.

