O artigo “Confined ionic liquids films under shear: The importance of the chemical nature of the solid surface” sob autoria minha e do Prof. Dr. Mauro C. C. Ribeiro foi publicado no Journal of Chemical Physics e pode ser acessado pelo endereço https://aip.scitation.org/doi/10.1063/5.0141388 .
Nesse trabalho usamos o campo de força coarse grained Martini 3.0 para simular o líquido iônico tetrafluoroborato de 3-metil-1-etil-imidazólio confinado entre duas superfícies sólidas e induzimos o cisalhamento fazendo essas superfícies se moverem em direções opostas. Propusemos nesse trabalho duas definições distintas para a viscosidade, a taxa de cisalhamento e a tensão de cisalhamento do líquido confinado: Uma definição local, baseada em propriedades do próprio líquido, e uma de engenharia, baseada em grandezas medidas nas próprias superfícies. Variamos a natureza da superfície sólida por meio dos parâmetros de interação com o líquido e demonstramos que interações fortes seja com o cátion ou com o ânion induzem cristalização na camada de líquido em contato com a superfície o que afeta o comportamento reológico do líquido.
Foi publicado recentemente na Science um artigo com colaboração teórico-experimental referente a formação e a agregação de nanoestruturas quirais formadas por ouro e cisteína. Minha colaboração para esse trabalho constitui na elaboração e uso de um modelo coarse grained para a realização de simulações dessas estruturas.
O vídeo abaixo mostra a secagem de uma suspensão de nanopartículas recobertas com ácido oleico em clorofórmio sobre uma superfície sólida utilizando simulações de dinâmica molecular com modelos coarse grained. São mostradas simultaneamente duas representações, uma com ênfase no espalhamento da suspensão sobre a superfície sólida e a evaporação do solvente enquanto a outra representação mostra as nanopartículas se reorganizando sobre a superfície mediante o processo de secagem.
Essa simulação foi parte de um trabalho publicado no PCCP em 2015 com autoria de André F. de Moura, Kalil Bernardino, Cleocir, J. Dalmaschio, Edson R. Leite e Nicholas A. Kotov com o título “Thermodynamic insights into the self-assembly of capped nanoparticles using molecular dynamic simulations” e DOI 10.1039/C4CP03519D , onde também foram apresentados resultados de potencial de força média para discutir a termodinâmica de associação das nanopartículas em suspensão e no vácuo. Uma descrição detalhada do processo de secagem e a comparação da estrutura obtida por simulação com imagem de TEM (microscopia de transmissão eletrônica) podem ser encontradas no artigo.
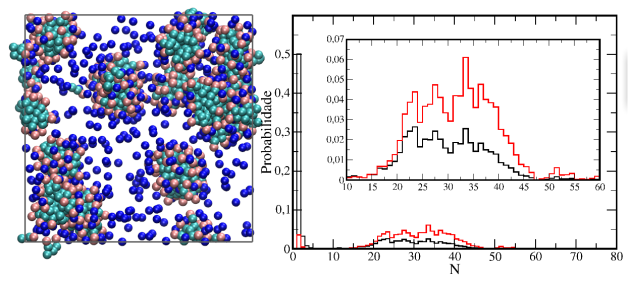
Acabo de adicionar ao blog a primeira de uma sequência de páginas sobre surfactantes. Essas páginas terão enfoque fenomenológico, com discussões focadas principalmente em aspectos estruturais dos agregados formados onde são apresentados resultados obtidos por meio de simulações de dinâmica molecular empregando modelos coarse grained.
Nessa primeira página é dada uma introdução discutindo aspectos gerais sobre surfactantes, como a estrutura típica desses e suas aplicações, e apresentados resultados para uma simulação de um surfactante aniônico em água na qual é observada a formação espontânea de micelas. São apresentadas e discutidas análises de distribuição radial de pares e de distribuição de tamanhos de agregados.
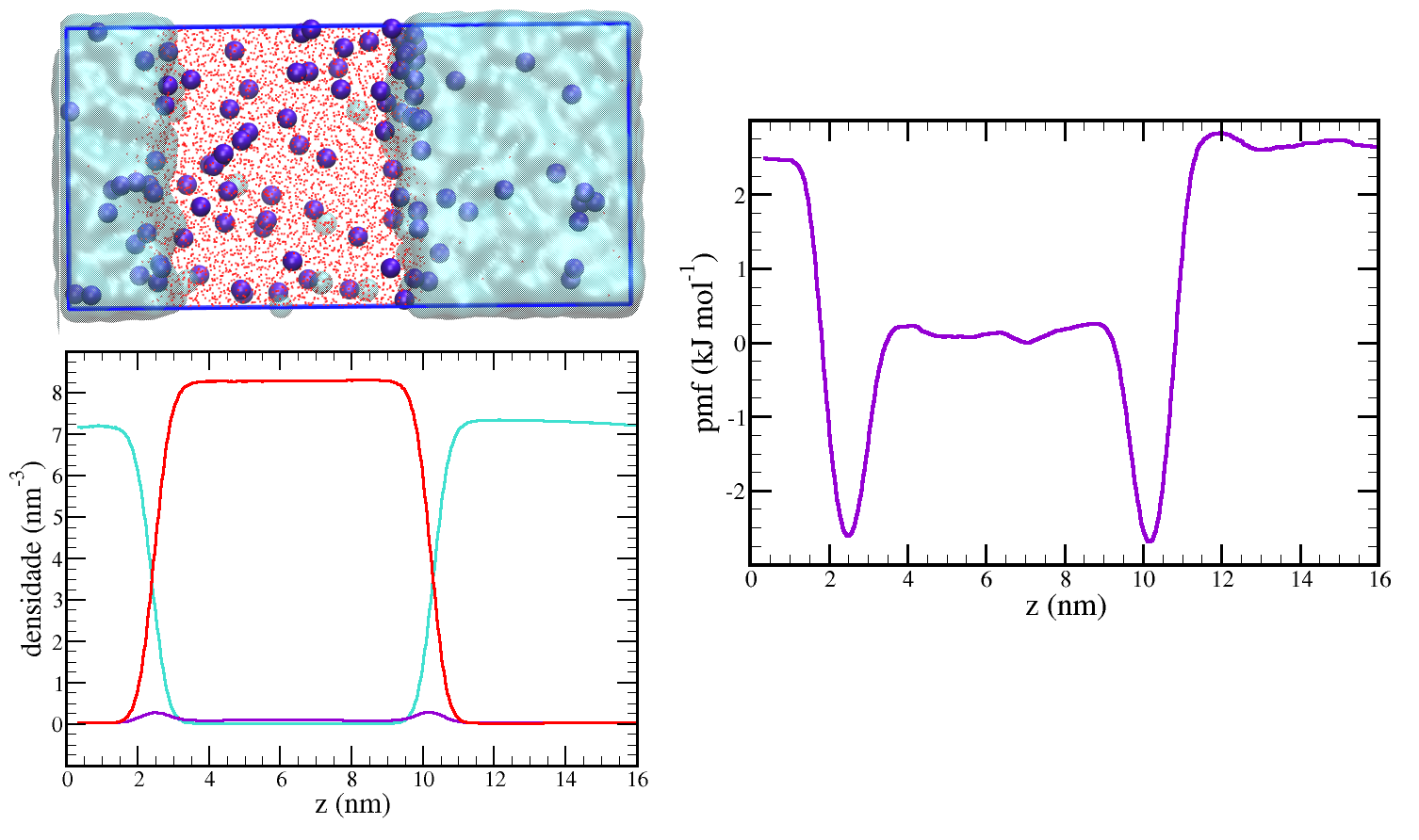
Nessa página é apresentado um estudo usando modelos coarse grained para sistemas com 2 ou 3 componentes líquidos nos quais ocorrem separação de fases. Nesse tipo de sistema é simples a obtenção do potencial de força média (pmf), que fornece o trabalho reversível necessário para mover uma molécula ou mesmo um agregado molecular de um ponto a outro no sistema. Em alguns outros sistemas a obtenção do pmf é dificultada por problemas de amostragem e técnicas especiais são necessárias para sua construção, sendo que algumas dessas serão abordadas futuramente em outros tutoriais de dinâmica molecular, para os sistemas descritos nessa página, o potencial de força médio pode ser construído através das distribuições de densidades médias do sistema obtidos através de uma simulação no equilíbrio.
Além do cálculo do pmf, discutiremos também sua interpretação física em termos da força média sentida pela partícula e sua correlação com conceitos mais familiares aos químicos como a solubilidade e a energia livre de adsorção em uma interface.
Em solução ou no estado fundido as moléculas de polímeros usualmente assumem uma conformação denominada novelo aleatório, na qual a molécula se enovela e apresenta um movimento browniano devido às colisões aleatórias das moléculas de solvente com ela. Além das colisões com as moléculas de solvente resultarem em movimentos de translação e rotação do polímero, como em um dado instante fragmentos distintos da cadeia podem sentir forças de intensidade e direção distintas, a molécula muda constantemente de conformação.
O quanto uma molécula de polímero pode extender-se em solução, ou seja, a máxima distância cabeça-cauda, definida com a distância entre as extremidades da molécula, depende obviamente da quantidade de unidades monoméricas e dos comprimentos das ligações químicas presentes no polímero. Em solução concentrada, a determinação da distância cabeça-cauda média exibida pelo polímero pode ser bastante complicada devido à presença de interações entre as diversas moléculas de polímero, mas no limite da diluição infinita a conformação, e portanto a distância cabeça-cauda média, dependem, além da quântidade de monômeros na molécula e dos parâmetros ligantes (comprimentos de ligação e ângulos de ligação e de diedros), apenas das interações não-ligantes entre diferentes segmentos da cadeia e entre as unidades monoméricas e o solvente, de modo que quanto mais intensa as interações monômero-solvente forem em relação às interações monômero-monômero e solvente-solvente, mais aberto tende a ser o novelo formado e maior a distância média cabeça-cauda.
Abaixo é mostrado um vídeo de simulações coarse-grained de 3 polímeros distintos em água usando o pacote computacional GROMACS 4.5.5 e o campo de força MARTINI 2.1 (veja mais detalhes nessa página, onde foram usadas os mesmos parâmetros de simulação). Cada simulação foi iniciada com a molécula de polímero esticada em uma caixa com 22414 partículas de solvente e foi observada a relaxação expontânea das cadeias. No campo de força MARTINI, cada sítio de interação representa 4 átomos pesados, sendo que foram construídos polímeros usando os sítios:
P4 – fortemente polar e prótico, algo como a unidade [-CH2-CHOH-] do poli-álcool vinílico (PVA). Como esse é o mesmo tipo de sítio usado para o solvente, essa simulação representa uma solução ideal do polímero, ou seja, com entalpia de mistura nula pois não há preferência para o polímero interagir com ele próprio ou com o solvente
N0 – polar e aprótico, como a unidade éter [-CH2-CH2-CH2-O-] no polióxido de propileno (POP). Nesse caso, as interações polímero-polímero são iguais às interações polímero-solvente, mas mais fracas que as interações solvente-solvente.
C1 – Completamente apolar, como cada sítio C1 corresponde a aproximadamente 4 átomos de carbono, cada sítio corresponde a duas unidades monoméricas [-CH2-CH2-] do polietileno (PE), por exemplo. Como esse é um polímero completamente hidrofóbico, espera-se que esse se enovele em água na forma mais compacta possível, como de fato ocorre.
Note que o solvente foi excluído no vídeo para melhor visualização e que, na verdade, foram feitas 3 simulações distintas, cada uma empregando um dos parâmetros de interação listados e as três trajetórias foram sobrepostas para facilitar a comparação simultânea no vídeo, então, onde se vê dois polímeros praticamente se tocando, isso é apenas um efeito da montagem feita com as trajetórias. Cabe ressaltar também que usando um modelo com 25 sítios, o que corresponde a 25 monômeros no caso do POP ou 50 no caso do PE, estamos longe do tamanho real de um polímero, que contém milhares de unidades monoméricas, mas os resultados obtidos demonstram as tendências esperadas: O polímero polar e prótico interage bem com a água e sua cadeia permanece a maior parte do tempo bastante estendida e exposta ao solvente. O polímero menos polar e aprótico, por outro lado, apresenta a cadeia parcialmente enovelada na maior parte do tempo, e o polímero apolar se enovela em uma forma aproximadamente esférica de modo a reduzir ao máximo sua exposição ao solvente.
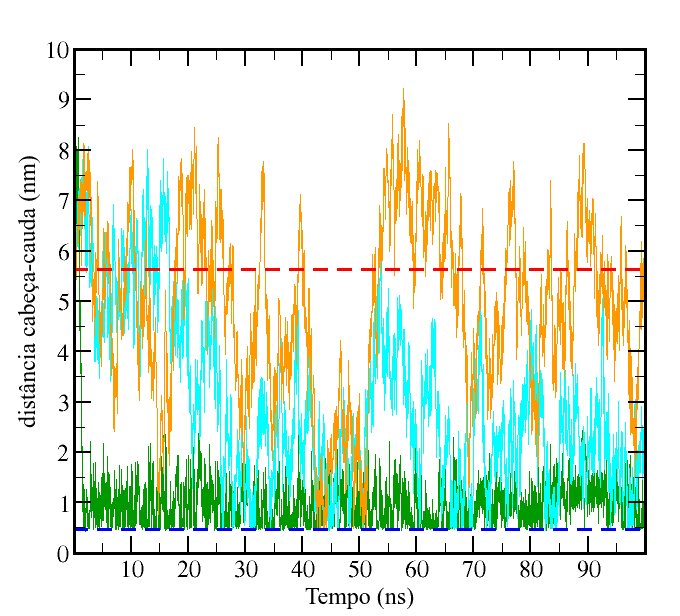
Abaixo, é mostrada a variação da distância cabeça-cauda dos 3 polímeros com o tempo, calculada com o programa g_mindist do GROMACS, onde cada curva tem a mesma cor usada para o respectivo polímero no vídeo: laranja para P4, azul para N0 e verde para C1. As distâncias de ligação entre os sítios coarse-grained e o parâmetro σ do potencial de Lennard-Jones para as interações não-ligantes valem ambos 0,47 nm, assim, essa pode ser tomada como a distância de contato cabeça-cauda e é representada pela linha azul tracejada. Em nenhum instante observa-se a distância cabeça-cauda no valor de extensão máxima com as ligações no comprimento de equilíbrio que é dada por 24 x 0,47 = 11,28 nm, então, como outra referência foi tomado o valor de 5,64 nm, metade dessa extensão máxima, representado pela linha tracejada vermelha. Note que o polímero P4 apresenta em boa parte do tempo distância cabeça-cauda superior a esse valor, enquanto o N0 raramente apresenta e o C1, após a rápida relaxação inicial, apresenta apenas valores inferiores.
distância cabeça-cauda para os três polímeros estudados. As cores das curvas são as mesmas usadas no vídeo.
Os valores médios com os respectivos desvios para a distância cabeça-cauda, calculados considerando os 50 ns finais da trajetória, são: (1,0 ± 0,3) nm para o polímero C1, (2,4 ± 1,1) nm para o N0 e (5,2 ± 1,6) nm para o P4.
Acabo de adicionar a primeira página do blog sobre simulações de dinâmica molecular empregando modelos coarse grained, que são modelos grosseiros nos quais diversos átomos são agrupados em um único sítio de interação. Nessa página é apresentada uma visão geral do campo de força MARTINI, que será empregado nas simulações coarse grained nesse blog e são discutidas simulações de líquidos puros, empregando potenciais que descrevem a água, álcoois, éteres e cetonas. O modelo apresentado será utilizado posteriormente no estudo de sistemas mais complexos, como misturas de líquidos pouco solúveis, polímeros em solução e microemulsões pois permitem a realização de simulações muito mais e com sistemas maiores do que as simulações atomísticas apresentadas até então, apesar da perda de detalhes em nível molecular.
As simulações apresentadas nessa página têm como objetivo principal demonstrar como o potencial de interação intermolecular, que é particularmente simples nos sistemas discutidos, afeta propriedades macroscópicas como a densidade e a entalpia de vaporização de líquidos, além da organização molecular do sistema.
Micela de SDS estudada por simulação de dinâmica molecular atomística
Micelas de SDS e octano formadas em uma simulação de dinâmica molecular
Blog com diversos exemplos de aplicações de Química Computacional criado por Kalil Bernardino, professor adjunto no Departamento de Química da Universidade Federal de São Carlos
Formação:
- 2005 - 2007: Ensino médio e técnico em informática no Colégio Network.
-2008 - 2011: Graduação no curso de Bacharelado em Química na Universidade Federal de São Carlos.
-2011 - 2013: Mestrado em Físico-Química na Universidade Federal de São Carlos com orientação do Prof. Dr. André Farias de Moura
- 2013 - 2018: Doutorado em Físico-Química na Universidade Federal de São Carlos com orientação do Prof. Dr. André Farias de Moura.
- 2018 - 2022: Pós-doutorado na Universidade de São Paulo sob supervisão do Prof. Dr. Mauro C. C. Ribeiro