Acabo de adicionar a segunda página do tutorial atualizado do Gromacs para a simulação de uma caixa de água. Nessa segunda parte focaremos nas análises da trajetória obtida, incluindo análises de ligações de hidrogênio, distribuição radial de pares e distribuição espacial. Além de ferramentas do próprio Gromacs, usaremos também o programa Travis para as análises.
Clique no link ou na imagem abaixo para ir à página:
(English below) A versão final do meu artigo sobre efeitos de pressão e cisalhamento na viscosidade e estrutura de líquidos iônicos estudados por simulação de dinâmica molecular de não equilíbrio está disponível! Em aplicações como lubrificantes, um filme líquido pode estar sujeito simultaneamente à altas pressões e taxas de cisalhamento. Nesse trabalho, 2 líquidos iônicos que diferem no tamanho do grupo alquil do cátion foram estudados utilizando modelos polarizáveis em diferentes condições de pressão e cisalhamento. Observou-se que pressões elevadas não apenas aumentam a viscosidade como fazem o comportamento não-newtoniano começar em taxas de cisalhamento mais baixas. Enquanto a pressão torna as camadas de coordenação mais bem definidas, o cisalhamento as torna mais difusas. Para efeitos de comparação, o mesmo estudo foi também conduzido para o benzeno. O link abaixo permite acesso gratuito ao mesmo até 22 de janeiro! ———————– The final version of my manuscript regarding the pressure and shear rate effects over the viscosity and structures of ionic liquids studied by means of non-equilibrium molecular dynamics simulations is available now! In applications as lubricants, a liquid film may be subject simultaneously to high pressures and high shear rates. In this work, two ionic liquids with different alkyl groups bonded to the cation were studied using polarizable models in different conditions of pressure and shear. High pressures not only increase the zero shear viscosity but also makes the non-newtonian behavior starts at smaller shear rates. While the pressure renders the coordination shells more defined, the shear rate makes them more diffuse. For comparison purposes, the same study was also performed for benzene. The link below gives free access to the paper until January 22!
Bernardino, K., Ribeiro, M. C. C. “Pressure and shear rate effects on viscosity and structure of imidazolium-based ionic liquids” Fluid Phase Equilibria 554 (2022) 113345.
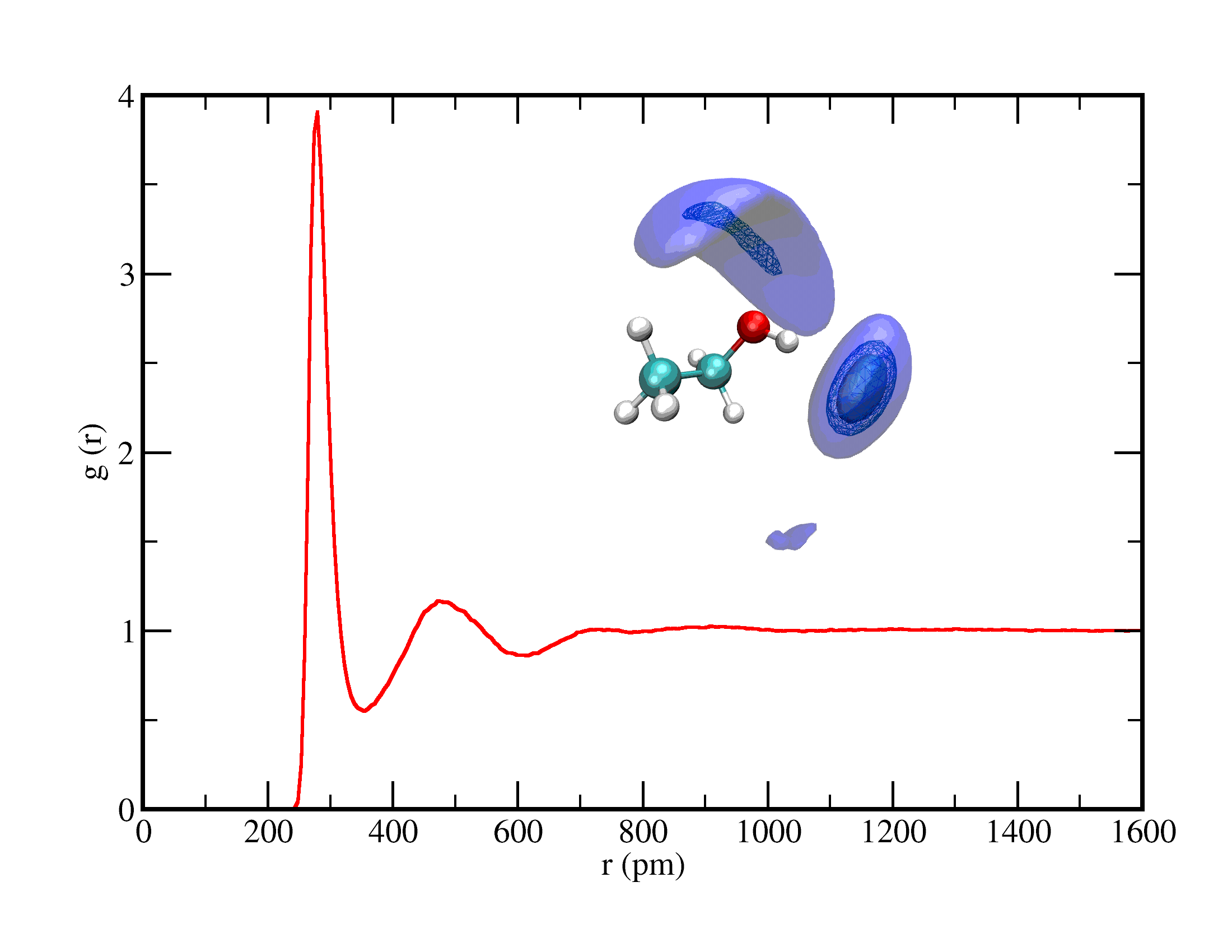
Acabo de adicionar a segunda página do tutorial do LAMMPS referente a simulação de uma mistura equimolar de água e etanol. Nessa página são abordadas análises de componentes de energia e densidade e análises estruturais como distribuição radial de pares, distribuição espacial e distribuição de diedros. Para acessar essa página, clique aqui ou na figura abaixo.
Distribuição radial de pares entre oxigênio da água ao redor de oxigênio de etanol na mistura equimolar e distribuição espacial de oxigênio da água ao redor da molécula de etanol.
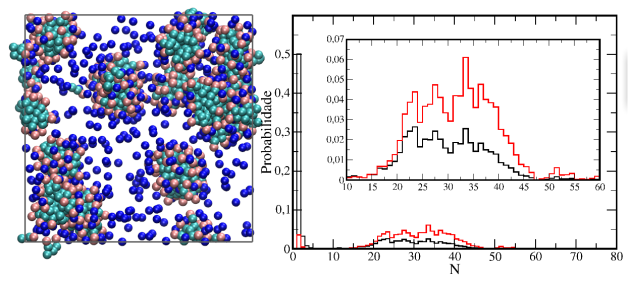
Acabo de adicionar ao blog a primeira de uma sequência de páginas sobre surfactantes. Essas páginas terão enfoque fenomenológico, com discussões focadas principalmente em aspectos estruturais dos agregados formados onde são apresentados resultados obtidos por meio de simulações de dinâmica molecular empregando modelos coarse grained.
Nessa primeira página é dada uma introdução discutindo aspectos gerais sobre surfactantes, como a estrutura típica desses e suas aplicações, e apresentados resultados para uma simulação de um surfactante aniônico em água na qual é observada a formação espontânea de micelas. São apresentadas e discutidas análises de distribuição radial de pares e de distribuição de tamanhos de agregados.
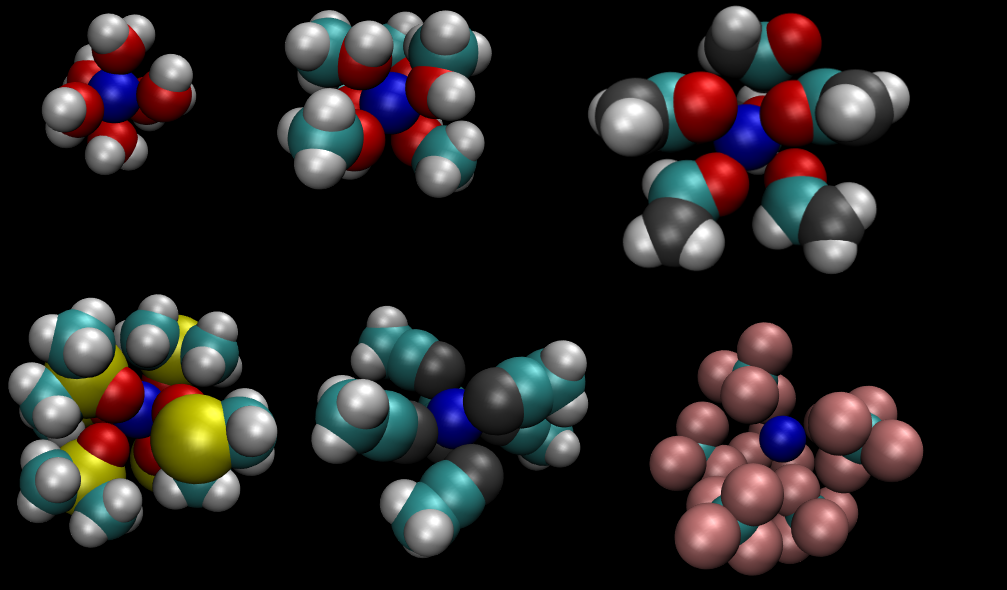
Acabo de adicionar a página sobre íons em solventes orgânicos, na qual são discutidos resultados de simulações de dinâmica molecular dos íons sódio e cloreto em soluções diluídas dos solventes metanol, formamida, DMSO, acetonitrila e tetracloreto de carbono, além de resultados em soluções aquosas para efeito de comparação.
Clique no link ou na imagem abaixo para ir à página:
íon sódio com sua primeira camada de solvatação em seis solventes distintos
Nessa página são discutidos resultados de energia de interação e distribuição radial de pares e são fornecidas as topologias e caixas de simulação dos solventes discutidos. Para mais detalhes sobre a realização das simulações, estrutura dos arquivos de input e sobre as ferramentas de análise empregadas, consulte os seguintes tutoriais de dinâmica molecular:
Estou adicionando uma página sobre estados-físicos da matéria, em que são apresentados resultados de simulações de argônio nos estados sólido, líquido e gasoso, além do estado metaestável de líquido super-resfriado e de fluído supercrítico, sendo comparadas diferenças energéticas e estruturais de cada estado além do comportamento dos diferentes estados em relação à temperatura. Essa página tem como principal objetivo evidenciar os diferentes estados físicos e demonstrar a utilidade de simulações computacionais para esse objetivo, sem a intenção de servir como um tutorial de dinâmica molecular, como foi o caso das páginas de dinâmica molecular postadas até o momento, mas serão fornecidos arquivos de input para a realização das simulações apresentadas.
Trata-se de um tutorial de duas páginas de dinâmica molecular onde é discutida, passo a passo, uma simulação de uma caixa com água utilizando o GROMACS, um pacote de aplicativos gratuíto para realização e análise de simulações de dinâmica molecular. Esse tutorial aborda os parâmetros de uma simulação de dinâmica molecular, os comandos e arquivos básicos do programa, softwares para visualização de estruturas e análises de componentes de energia, densidade, ligações de hidrogênio e distrbuição radial de pares.
Micela de SDS estudada por simulação de dinâmica molecular atomística
Micelas de SDS e octano formadas em uma simulação de dinâmica molecular
Blog com diversos exemplos de aplicações de Química Computacional criado por Kalil Bernardino, professor adjunto no Departamento de Química da Universidade Federal de São Carlos
Formação:
- 2005 - 2007: Ensino médio e técnico em informática no Colégio Network.
-2008 - 2011: Graduação no curso de Bacharelado em Química na Universidade Federal de São Carlos.
-2011 - 2013: Mestrado em Físico-Química na Universidade Federal de São Carlos com orientação do Prof. Dr. André Farias de Moura
- 2013 - 2018: Doutorado em Físico-Química na Universidade Federal de São Carlos com orientação do Prof. Dr. André Farias de Moura.
- 2018 - 2022: Pós-doutorado na Universidade de São Paulo sob supervisão do Prof. Dr. Mauro C. C. Ribeiro